Анализ структуры аутосомного генофонда популяции беларусов: результаты анализа этнического адмикса.
После проведения анализа этно-популяционного адмикса мы получили следущие результаты, обсуждению которых будет посвящена следущая часть нашего исследования. Результаты представляют собой разбивку аллельных частот на 22 кластера, каждый из которых представляет собой гипотетическую предковую популяцию. Поскольку в цели данного небольшого исследования не входит подробный анализ всех популяций, мы ограничимся сравнительном анализом структуры (компонентов) беларусов c географически близкими популяциями, а также с теми популяциями, которые могли входить в исторические контакты с предками современных беларусов:

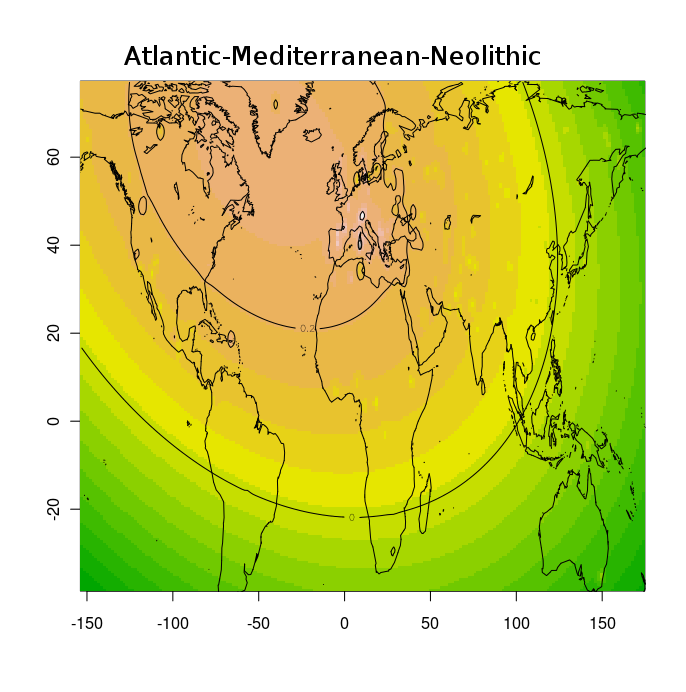
Рисунок 3. Результатыанализа ADMIXTUREK=22
У рассматриваемых здесь европейских популяций наиболее часто представлены следующие компоненты:
North-East-European,Atlantic_Mediterranean_Neolithic,North-European-Mesolithic, West-Asian, Samoedic, Near_East.
Разберем вкратце каждый из них. В ракурсе нашего исследования самым важным компонентом представляется – северо-восточно-европейский компонент North-East-European, он присутствует почти у всех европейцов, и в самой значительной степени — у балтов и славян: литовцы (81,9), латыши (79,5), беларусы (76,4), эстонцы (75,2), поляки (70,2), русские (67- 70,4), украинцы (62,1- 67,1), сорбы (65,9), карелы (60,2), вепсы (62,5), чехи (57,4), северные немцы (54,6), южные- 42,6, у британцев от 46 до 49, норвежцы- 48,1, шведы- (53,7).
Второй по значимости компонент — Atlantic_Mediterranean_Neolithic (юго-западно-европейский или просто западно-европейский неолитический компонент).[1]У восточноевропейцев он выражен в умеренной степени- чехи (27,8), поляки (18,4), украинцы ( от 17 до 21%), беларусы (13%), русские (от 11 у северных до 17,3 у южных), у коми (8,9 %), манси (8,8 %).
Третьй компонент – северо-европейский мезолитический компонент -North-European-Mesolithic[2]: cаамы (76,4 %), финны (от 30,1 до 37,3 %), вепсы (24,1), карелы (23,2), ижорцы (22, 7). Заметен этот компонент и у северных русских (10,5 %), норвежцев (9,8 %), шведов (7,8 %), эстонцев (7,1 %). У беларусов он практически отсутствует (1.1%).
Четвертый компонент – западно-азиатский (кавказский) West Asian[3]. На интересуемой нас территории этот компонент чаще встречается у казанских татар (9,9 %), южных немцев (8,4), украинцев (от 6,6 до 7,7 %), южных русских (6,2%). На западе высок процент у итальянцев (21,5 % у центральных итальянцев), французов (6,7 %), у беларусов (2.2%).
Пятый компонент — уральский Samoedic. Значительно присутствует у селькупов (68,1%), хантов (64,6), ненцы (37,1), манси (30,9 %-), удмурты (29,6), марийцы (27, 8), шорцы (22,0 %), башкиры (21,7%), чуваши и хакассы по 17,6 %, коми- 16,4 %, казанских татар (11,9 %). У западноевропейцев этот компонент практически не встречается, у русских (от 1,0% у центральных до 4,7 % у северных), у карел (1,6%), словаков (1,4%), западных украинцев (1,7 %), беларусы (0.5%).
Шестой компонент – ближневосточный Near_East[4]У южных немцев (3,5), украинцы (от 2,3 у восточных до 3,8 % у западных), чехи (3,0), беларусы (3,4), словаки (3,2), у русских от 1,0 до 1,5%, у литовцев- 1,4%, у поляков- 1,3 %.
[1]Больше всего у сардинцев (68,1 %), басков (59,2 %), иберийцев (48,8), корсиканцев (47,8), португальцев (46,6), северных итальянцев (44,3), французов (43,5 %). Данный компонент достаточно выражен у всех западноевропейцев- более 30 %
[1]Название связано с тем, что этот компонент достигает значительных частот в древней ДНК жителей мезолитической Иберии, неолитических жителей Швеции и современном ДНК жителей Фенноскандии
[1]Наибольший процент на Западном Кавказе- абхазы (64, 9%), имеретинцы (63,7), лазы (56,6), аварцы (56,8), лезгины (55,4).
[1]Евреи Йемена (60,9 %), Сауд. аравия (59,5), бедуины (56,7), евреи Эфиопии (52,5), египтяне (43,8).В Европе oтносительно много у итальянцев (центр- 17,4), португальцев (11,9).
Анализ разделяемых аутосомных сегментов между популяциями Северо-Восточной Европы.
С целью верификации результатов анализа главных компонентов генетического разнообразия я подготовил новую выборку популяций, которая включает в себя ряд референсных евразийских популяций и анализируемую группу участников моего проекта MDLP. В совокупности, выборка включала в себя 900 индивидов, каждый из которых был типирован по 350 000 снипам.В ходе нового экспериментального теста в ходе статистической обработки общих по генетическому происхождению сегментов хромосом в составе выборки было выделенно 15 групп кластеров генетически близких популяций Как нам представляется, ключевым моментом для понимания принципов этого анализа, а также результатов, является понятие эффективной популяции или эффективный размер (Ne) популяции, т.е размера той популяции которая участвовала в репродукции или обмене генами в некоем отдаленном временном промежутке. Собственно говоря, эффективная популяция – это даже не число уникальных предков, а математическая абстракция разброса гамет, размер которого оценивается исходя из разброса числа гамет относительного к гамет, передаваемых родителям репродуктивного возраста следующему поколению. Он отличается от репродуктивоного объема Nr в той мере, в какой существует неравный вклад лиц родительского поколения в генофонд следующего поколения. Это создает разброс значений числа гамет к, того родителя относительно числа гамет к, передаваемых родителям следующему поколению (Wright, 1931, Li Ch. Ch., 1955). Новая программа Chromopainter позволяет оценить этот размер, исходя из числа наблюдаемых рекомбинаторных гаплотипов и значений LD. Когда я производил оценку этого размера, то для каждой из 22 неполовых хромосом он получился разный, однако среднеарифметическое значение составило 22 000. Это близко к значениям Neрекомендованным к использованию профессионалами (например, авторами программы IMPUTE V2). Как видно из приведенных ниже результатов, даже 22 000 для совокупности эффективного размера элементарных популяций – это более, чем достаточно.

Рисунок 4. Расположение популяций в пространстве 1 и 3 главных генетических компонентов
Изложим ниже некоторые закономерности размещения популяци
- Финны оказались ближе к русским и поволжским финно-угорам (эрзя и мокша)
-
Все литовцы (участники проекта + референсы из вышеупомянутой статьи Бехара) и часть референсных белорусов образовали отдельный кластер, тесно примыкающий к кластеру белорусов, поляков, украинцев
-
Следущим кластером является центрально-европейский кластер, представленный главным образом венграми, хорватами, а также частью немцев.
-
Ниже находится балканский кластер (румыны, болгары и часть венгров).
-
К этому кластеру примыкают турки и часть армян
-
В центре плота находятся западноевропейцы из моего проекта (французы, немцы, бельгийцы и жители британских островов).
-
Выше находятся два оркнейских кластера, в которых находится и часть скандинавских сэмплов.
-
Еще левее находится кластер образованный референсными северо-итальянцами и тосканцами.
-
Ниже находятся армяне и слево итало-иберийский кластер (часть итальянцев и испанцы).
-
Левее этой группы популяций находится кластер ашкеназов.
-
Наконец, самый крайний слева кластер представлен изолированной популяцией сардинцев.
-
Ниже итало-иберийского и армянского кластеров расположен целый ряд кавказский кластеров. Это прежде всего адыгейцы и абхазцы, затем северные осетины.
-
Вышеназванные кластеры частично перекрывают кластер ногайцев (что свидетельствует о наличии генетического обмена между северокавказскими популяциями и ногайцами)
-
Кластер ногайцев плавно переходит в кластер узбеков, который в свою очередь примыкает к изолированному кластеру чувашей
-
Наконец самым изолированным кластером является кластер французских басков (в нижнем левом углу плота).[5]
[1]Больше всего у сардинцев (68,1 %), басков (59,2 %), иберийцев (48,8), корсиканцев (47,8), португальцев (46,6), северных итальянцев (44,3), французов (43,5 %). Данный компонент достаточно выражен у всех западноевропейцев- более 30 %
[2]Название связано с тем, что этот компонент достигает значительных частот в древней ДНК жителей мезолитической Иберии, неолитических жителей Швеции и современном ДНК жителей Фенноскандии
[3]Наибольший процент на Западном Кавказе- абхазы (64, 9%), имеретинцы (63,7), лазы (56,6), аварцы (56,8), лезгины (55,4).
[4]Евреи Йемена (60,9 %), Сауд. аравия (59,5), бедуины (56,7), евреи Эфиопии (52,5), египтяне (43,8).В Европе oтносительно много у итальянцев (центр- 17,4), португальцев (11,9).
[5]Такое поведение на плоте объясняется только изолированным положением популяции и небольшим числом эффективной популяции.То есть все эти баски являются многократными родственниками между собой т.е., положение басков на графике есть следствие классического генного дрейфа, который можно наблюдать на карте.На самом деле положение басков на данном плоте не может ни подтвердить, ни опровергнуть гипотезу о континуитете баскской популяции , т.к PCA-координаты (eigenvalues и eigenvectors) вычислялись в Chromopainter исходя из количества sharedDNAchunks между популяциями-донорами и популяциями-рецепиентами.То есть баски изоляты в том смысле, что уровень обмена ДНК между ними и другими популяцими ничтожен.
Исходя из этого можно сделать вывод о том что баски эта экстремально-эндогенная популяция изолянтов, при этом генетическое разнообразие басков низко, т.к. размер эффективной популяции басков низок.
















Для отправки комментария необходимо войти на сайт.