В марте прошлого года Сергей Козлов — один из соавторов данного блога, — опубликовал важную с точки зрения методологии генетико-генеалогического анализа заметку о принципах оценки вероятности определения времени жизни последнего общего предка при попарном сравнении аутосомных данных двух или более сравниваемых индивидов. Действительно, в последние годы среди людей, интересующихся генеалогией, приобрели заметную популярность сервисы, производящие поиск генетических родственников по всем линиям, а не только по прямой мужской и прямой женской. В качестве примера можно привести Family Finder от FTDNA и DNA relatives от 23andMe. Участник получает достаточно длинный список так называемых «совпаденцев» — людей, имеющих с ним один или более участок половинного совпадения (УПС) на аутосомах (неполовых хромосомах). Если участок достаточно длинный (а его длина измеряется в сантиморганидах, обозначающих вероятность разрыва участка при каждой передаче в следующее поколение), то это говорит о наличии общего предка (от которого участок и получен).
Для значительной части клиентов сервисов персональной коммерческой геномики, интересующихся исключительно вопросами своего происхождения, вопрос о достоверном определении времени жизни общих предков имеет первостепенное значение. И вместе с тем, именно проблема с получением четкого ответа на этот краеугольный вопрос служит одной из главных причин недовольства и раздражения клиентов компаний вроде FTDNA или 23andme.
Действительно, изучив длинные сегменты генома, передававшихся от поколения к поколению и встречающиеся у многих людей, можно примерно определить степень и интенсивность предковых связей, берущих начало много тысяч лет назад. Здравый смысл подсказыает — дальние родственники имеют такие длинные сегменты генома потому, что они унаследовали их от общих предков. У более далеких родственников длина сегментов общих геномов соответственно становится короче, поскольку происходит рекомбинация гомологичных хромосом, в результате чего с каждым следующим поколением происходит перемешивание всей совокупности генов или генотипа. Очевидно, что число и размер совпадающих общих по происхождению сегментов геномов у двоих произвольно взятых лиц из однородной метапопуляции коррелирует с географический дистанцией — количество общих генетических предков резко уменьшается по мере увеличения географического расстояния.
Однако наряду с географически близкими (в пределях 50-100 км) «совпаденцами», нередко в списках «совпаденцев», предоставляемых в 23andme или FTDNA появляются совершенно экзотические «совпаденцы». Например, у финна может появится совпаденец из Италии, а у корейца — из Великобритании. Совершенно очевидно, что подобные случаи очень сложно объяснить не только простым сопоставлением сведений о географическом происхождении предков, но даже и безотказной в простых случаях моделью наложения «этнопопуляционного аутосомного фона в виде коротких реликтовых IBD сегментов».
В этой связи возникает практический вопрос — как интерпретировать подобные случаи, при условии что подобные сегменты представляют собой не «ложно-позитивные», а вполне достоверные совпадения, указываюшие на существование в неопределенный момент прошлого некоего общего предка. И подобные случаи характерны не только для коммерческих «выборок», но и для вполне серьезных научных баз данных, например 1000 Genomes. В частности, в этой базе данных при сравнении редких снипов у 89 британцев и 97 китайцев были обнаружены три англо-китайские пары с отдаленным генеалогическим родством ( в геноме этих пар были обнаружены идентичные по происхождению фрагменты (IBD сегменты) ДНК, которые составляют 0,001%, 0,004% и 0,01% их геномов).
Самое простое решение этой проблемы некоторые из любителей генетической генеалогии пытались найти в обращении к сервисам главного инструмента аутосомной генетической генеалогии Gedmatch. В частности, как известно, данный сервер содержит онлайн-версии практически всех популярных среди любителей модификаций DIYDodecad калькуляторов. Например, выбрав разработанный мною калькулятор MDLP K23b в режиме Chromosome painting: Paint differences between 2 kits, 1 chromosome и сравнив характер распределения предковых компонентов на гомологичных хромосомах у двух сравниваемых людей, можно получить примерное представление о географическом ареале, в котором мог жить общий предок этих людей (вероятно, на этот ареал будет указывать доминирующий на совпадающем сегменте компонент). Логика простая. Предположим, например, что мы сравниваем сегменты хромосомы X в данных индивида A этнического происхождения D c данными индивида В этнического происхождения С. Здесь возможны три варианта
- С-происхождение предка или предков индивида A
- D-происхождение предка или предков индивида B
- Y-происхождение подмножества предков обоих индивидов
Используя эту логику, можно предположить что если в попарном сравнении сегмента обозначится хорошо выраженное преобладание (по отношению к средним значениям) компонента, характерного для этнопопуляции С, то следует выбрать первый сценарий; аналогично, если обнаружится избыток компонентов характерных для этнопопуляции D, то следует выбрать второй сценарий; если будет замечено преобладание редких для этнопопуляций С и D компонентов, то следует остановится на третьем варианте.
Пример I.
В этом примере мы будем использовать свои данные и данные женщины, с которой у нас был обнаружен подтвержденный генеалогией общий предок, живший в середине 19 века. При сравнении наших данных, алгоритм поиска достоверных генеалого-генетических совпадений обнаружил три сегмента с генетической дистанцией > 7 cантиморганов, cостоящих в блочной записи из более чем 700 последовательно совпадающих снипов
| Start Location | End Location | Centimorgans (cM) | SNPs | |
| 4 | 32232224 | 42421625 | 13.2 | 1115 |
| 7 | 8295405 | 13845989 | 9.8 | 885 |
| 11 | 36784445 | 45084878 | 8.0 | 881 |
Самый большой сегмент = 13.2 cM
Общий размер сегментов с сантиморганах > 7 cM = 30.9 cM
Приблизительное число поколений до общего предка = 4.4
Задетектированные сегменты хромосом идеографически отображаются при попарном сравнении в цветовой гамме — черный цвет означает несовпадающие сегменты, другие цвета — компонентную привязку к одному из компонентов моего калькулятора MDLP K23b. Ниже приведены фрагменты идеографического отображения 2 из 3 вышеуказанных совпадающих сегментов на кариограмму 4 и 7 хромосомы.:


Самый значительный сегмент (13.2 сM) на 4 хромосоме имеют хорошо заметную привязку к северо-восточно-европейскому компоненту [зеленый цвет], в исторической перспективе связанному с наследием мезолитического населения этого региона. А вот сегмент на 7 хромосоме имеет более сложную структуру, в которой характерно преобладание кавказского компонента [голубой цвет]. Таким образом можно уверено утверждать, что общий предок (или предки) могли жить в регионе восточной Европы.
К сожалению, данный инструмент сегментного сравнения на Gedmatch хотя и прост в обращении (в силу интуитивной понятности), однако далек от совершенства. В первую очередь, на аккуратность определения «генографического»происхождения сегмента влияет отсутствие на сервере гаплоидных фаз похромосомных данных. В результате, сравнение ведется не по конкретной фазе (т.е по конкретной хромосоме доставшейся ребенку от каждого из родителей), а по диплоидному составному блоку, т.е вместо настоящих IBD мы можем оперировать half-IBD (HBD), которые на слэнге русскоязычных любителей именуются УПС-ами. Во вторых, аккуратность генографического определения зависит от аккуратности определения предковых компонентов в используемом варианте калькулятора, но это отдельная тема для разговора.
К счастью, парадокс «экзотических» совпаденцев имеет более точное решение с помощью одной из программ, позволяющих определять геногеографическую структуру или «локальное происхождение» совпадающих сегментов. Можно использовать разные программы, HAPMIX, LAMP , HAPAA, ANCESTRYMAP — так как несмотря на ряд принципиальных отличий, все они используют алгоритмы моделнй скрытых марковских цепей (HMM) и поэтому выдают в целом схожие результаты. К этому же классу программ относится и более новая програма PCAdmix, которую я буду использовать в своем втором примере, в котором я задействую фазированные в BEAGLE генотипы. В целях разжевывания принципов работы программы, следует вкратце описать рабочий процесс PCAdmix.
PCAdmix являет cобой метод, который оценивает локальное происхождение хромосомных сегментов с помощью анализа главных компонентов (PCA) фазированных гаплотипов. В самом начале выполняется анализ главных компонентов в 2-3 референсных панелех, необходимых доя построения пространства главных компонентов, например, для хромосомы 22 . Поскольку метод использует фазированные данные, каждая копия хромосомы 22 в референсных панелях рассматривается как отдельная точка в пространстве главных компонентов. Первые две главные компоненты, как правило, представляют собой оси «предкового» расхождения популяций референсных панелей, что хорошо заметно на графиках. Если подобного рассхождения не наблюдается, то скорее всего в популяциях референсных панелей «маскируется» присутствие неявной популяционной субструктуры. В построенное таким способом пространство главных компонентов в дальнейшем проецируется группа лиц «смешанного» происхождения, и затем определяется значение нагрузки главных компонентов для каждого снипа. После этого метод переходит к анализу коротких «окон» снипов — для каждого из этих окон вычисляются вероятности того, что данное окно в гаплотипе человека «смешанного» происхождения происходит от одной из референсных популяций. Вычисленные таким образоом вероятности различных вариантов происхождения каждого окна снипов, используются на заключительном этапе метода в скрытой моделе Маркова (HММ) для сглаживания шума в определении происхождения «окон» снипов. Таким образом, данная скрытая модель Маркова НММ зависит от значений главных компонентов, доли каждого «компонента происхождения» на заданной хромосоме, а также матрицы перехода, которая, в свою очередь, зависит от числа поколений прошедших с момента смешивания популяций и генетического расстояния (сM) между двумя окнами снипов. В текущей версии метода, рекомбинаторные расстояния и число поколений определяются параметрами.
Конечным результатом рабочего процесса PCAdmix является матрица состяний скрытой модели Маркова, содержащая апостериорную вероятность каждого из возможных вариантов происхождения для данного «окна снипов», и эта вероятность обусловлена остальной частью данных для хромосомы. Важно отметить, что происхождение каждого окна снипов определяется только в том случае если апостериорная вероятность для одного из возможных происхождений > = 0,8. Любое окно, для которого максимальная апостериорная вероятность любого варианта происхождения <0,8, считается «неопределенным».
Пример 2
Данный пример основан на реальном случае, когда ко мне обратился человек, чьи предки происходят из центральных регионов Азии. Смущенный наличием в списке своих совпаденцев в сервисе Relative Finder 23andme человека с корейскими и японскими корнями, а также семейными легендами о «восточноазиатской»прабабушке, он попросил меня определить вероятность присутствия японцев в числе своих ближайших (в пределах 5 поколений) предков, опираясь исключительно на аутосомные данные.
В этом эксперименте, я решил скурпулезно следовать инструкциям разработчиков PCAdmix, и для начала произвел фазирование (биоинформатическую реконструкцию гаплотипных фаз аутосомных хромосом) в программе BEAGLE. Данные тестанта (ок 400 тыс. снипов) были фазированы в присутствии 3 контрольных референсных групп популяций — британцев GBR, китайцев CHB и японцев JPT — поскольку эти группы были позднее задействованы мной в качестве 3 референсных панелей. В целях уменьшения количества ошибок, которые неизбежно появляются в результате импутации пропущенных «генотипов» снипов, я использовал только те общие снипы, которые были определены как в аутосомных данных клиента 23andme, так и в трех референсных группах.
Затем фазированные данные тестанта были похромосомно обработаны в рабочих циклах программы PCAdmix. Программа отфильтровала cнипы с низким значением MAF и высоким значением LD, в результате чего число снипов уменьшилось почти вдвое. Оставшиеся снипы были разбиты на «окна снипов», каждое из которых состяло из 20 снипов. При расчете по всем 22 хромосомах, общее количество полученных таким разбиением «окон» составило 11 997. В конце рабочего цикла (метод главных компонентов + HMM) программа выдала для каждой парной аутосомной хромосомы A и B файл в формате bed, удобном для отображения дополнительной информации в аннотации генома (номер хромосомы, начало и конец сегмента, наиболее вероятный регион происхождения сегмента, cM, максимальная вероятность и апостериорная вероятность одного из трех вариантов происхождения — JPT, GBR, CHB, непоказана в таблице). В конечном отчете GBR используется как индикатор сегментов не-восточноазиатского происхождения (nEA), JPT — японского происхождения (JPA), CHB — неспецифичных сегментов восточноазиатского происхождения (EA) :
| 10 | 111955 | 468599 | GBR | 0.004885 | 0.134147 | GBR* | 0.636943 |
| 10 | 521723 | 811876 | GBR | 0.142147 | 0.582463 | GBR* | 0.646868 |
| 10 | 815149 | 1151723 | GBR | 0.585829 | 0.898724 | GBR* | 0.676252 |
| 10 | 1156487 | 1335849 | GBR | 0.901503 | 1.23673 | GBR | 0.925059 |
| 10 | 1337709 | 1449849 | GBR | 1.24246 | 1.60705 | GBR | 0.99999 |
| 10 | 1454864 | 1510208 | GBR | 1.61249 | 1.76798 | GBR | 0.999506 |
| 10 | 1512546 | 1623734 | GBR | 1.77039 | 2.12653 | GBR | 0.999647 |
| 10 | 1624900 | 1669347 | GBR | 2.13038 | 2.25357 | GBR | 0.999778 |
Выбор формата BED в качестве формата выходных в моем случае также был далеко неслучайным. C помощью одной из библиотеки платформы Bioconductor формат BED легко отображается в кариограмме 22 пар аутосомных хромосом человека (я использовал координаты геномного билда b37). Чтобы было понятно, что именно изображают эти «кариоплоты» (идеографические изображения хромосом), необходимо пояснить, что «японское происхождение» (JPA) приписывалась 20-сниповому сегменту только в том случае, если апостериорная вероятность японского происхождения данного «окна из 20 снипов» составляла > = 0,8. Любое окно, для которого максимальная апостериорная вероятность любого варианта составляля <0,8, засчитывалось как окно с «неопределенным» происхождением (UND).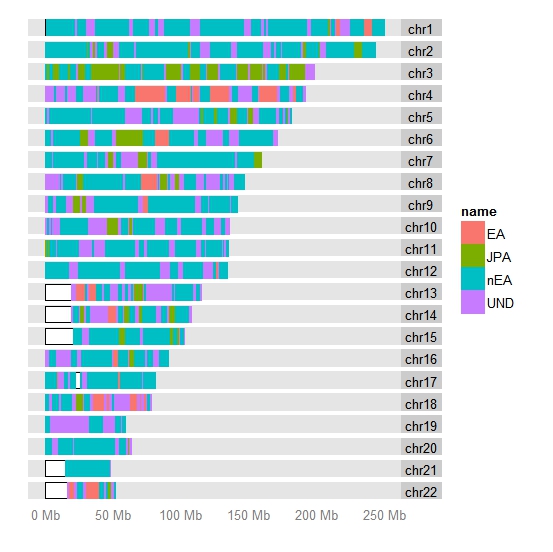
Chromosomes A

Эксперимент показал, что среди 11997 «окн» число «окон» не-восточноазиатского (nEA) происхождения (7650) почти в два раза больше чем число «восточноазиатских» сегментов. Происхождение 2750 геномных «окон» снипов невозможно определеить, и только 965 «окна» могут быть определены как «японские по происхождению». Вместе с 617 окнами «китайского» (EA), восточно-азиатские сегменты составляют меньше, чем 10% генома.
Не менее важно и то обстоятельства, что значительная доля этих сегментов-окон пришлась на низких «консервативные, низкорекомбинантные» области хромосом, — такие, как например, теломеры, центромеры и регионы с низкой плотностью снипов: сегменты в таких регионах могут переходить от одного поколения к другому фактически в неизменном виде. Наконец, те же закономерности распределения родословной были отмечены в обеих фазированных наборах аутосомных хромосом, что опровергает версию о недавной «восточноазиатской» примеси со стороны одного из родитедей и скорее свидетельствует о древнем эпизоде смешивание определенных центрально- и юго-западноазиатских групп с группами восточноазиатского происхождения (например, в ходе монгольских или тюркских нашествий).
Разумеется, как и во многих других моделях анализа, основанных на вероятностях, наше заключение нельзя считать окончательным вердикторм. Вместо этого, лучше сказать, что шансы в пользу существования «недавнего японского предка» против шансов отсутствия такого, составляют 10 к 90. Другими словами, вариант с недавней японской «примесью» нельзя полностью исключить, поскольку вероятность такого сценария составляет 11%.


















 )
)














{kind=link}
Для отправки комментария необходимо войти на сайт.