Сергей Козлов
Структура генофонда населения Русского Севера по аутосомным данным
Оригинал статьи расположен на ресурсе генофонд.рф
Содержание:
- Цель работы, применяемые методы и инструменты.
- Использованные выборки и источники их формирования. Некоторые важные работы, рассматривавшие тему северного генофонда.
- Основные компоненты аутосомного генофонда северян.
- Анализ взаимосвязей между неславянскими народами Севера.
- Структура аутосомного генофонда северных русских (включая Урал и Сибирь).
Цель работы, применяемые методы и инструменты.
Цель данной работы – проанализировать то, как сложился аутосомный генофонд северных русских (до массовых миграций XX века), из каких компонентов он состоит и что послужило их источником. Для решения этой задачи создана модель, совместно использующая два подхода – IBD-анализ и сравнение пропорций компонентов Admixture. Метод экспериментальный и не претендует на то, чтобы служить истиной в последней инстанции. И все же, на мой взгляд, он позволяет улучшить детализацию и разглядеть дополнительные подробности по сравнению с полученными ранее результатами. По мере пополнения новыми данными модель может изменяться, или же послужить основой для других моделей и интерпретаций.
IBD-анализ – это подсчет количества достаточно длинных общих участков ДНК, полученных от общего предка (IBD-сегментов) между образцами из исследуемых выборок. Многие используемые при аутосомном анализе методы позволяют определить, насколько сравниваемые образцы схожи или различны между собой, но не дают прямого ответа – является ли обнаруженное сходство свидетельством исторически недавнего родства?
Например, находящиеся рядом на графике главных компонент образцы могут относиться к разошедшимся многие тысячелетия назад популяциям, или же сходство может вообще оказаться результатом конвергенции (когда сумма внешних влияний на сравниваемые популяции схожа, но приведшие к ним события происходили совершенно независимо друг от друга). И наоборот, происходящие от общей основы, но испытавшие сильно различающиеся влияния популяции окажутся на графике далеко друг от друга.
Именно для решения этой проблемы наиболее пригоден IBD-анализ. Поскольку при каждой передаче следующим поколениям часть общих сегментов укорачивается или теряется, их суммарная длина и количество находятся в прямой зависимости от родственности между популяциями. К сожалению, использовать это как непосредственный показатель уровня родства нельзя, поскольку результат очень зависит от популяционной истории – в первую очередь, от испытанного популяцией генетического дрейфа и снижения разнообразия. Например, у эстонцев сумма общих сегментов с восточнофинской выборкой больше, чем с собственно эстонской, поскольку финны испытали более сильный дрейф. Поэтому был применён более сложный подход. На первом этапе среди исследуемых популяций выделяются те, которые испытали наиболее значительный дрейф и родство с ними хорошо проявляется по IBD-сегментам. Такие выборки называются у меня «генетическими полюсами». Далее выделяются опорные популяции, которые будут служить основой для моделирования (остальные выборки представляются, как результат их смешения). К опорным выборкам могут относиться как «генетические полюса», так и другие. Например, выборки центральных и северо-западных русских не формируют «генетических полюсов», но несомненно нужны в качестве опорных при моделировании происхождения северных русских.
Для поиска наиболее хорошо подходящих комбинаций опорных выборок использован алгоритм, называющийся «оракул» (насколько мне известно, впервые примененный геномным блогером Dienekes Pontikos). Он перебирает варианты смешения отобранных выборок и ищет результаты, где среднеквадратичное отклонение от целевой выборки будет наименьшим. В данной работе вклад каждой выборки при переборе изменяется с шагом 5%, поскольку шаг в 1% увеличит количество вычислений на порядки, но не даст реального повышения точности. Оракул может использовать как результаты IBD-анализа (тогда в качестве сравниваемых показателей используются суммы общих сегментов опорных выборок с «генетическими полюсами»), так и пропорции компонентов Admixture. Здесь они выделяются согласно разработанной В. Вереничем 27-компонентной модели, которую я считаю наиболее удобной для сравнения восточноевропейцев между собой, при помощи инструмента DIYDodecad от Dienekes. Что касается IBD-сегментов, то выбраны следующие показатели – чтобы исключить случайные совпадения, длина каждого учитываемого сегмента должна быть не менее 3 сМ, и он должен состоять не менее, чем из 150 снипов. При сравнении двух выборок итоговая цифра показывает усредненный результат сравнения каждого генома из первой выборки с каждым геномом из второй (сумма в сМ).
Программные реализации оракула, IBD-анализа и алгоритма отображения на картах, применяемые в данной статье, созданы непосредственно автором.
Использованные выборки и источники их формирования.
В статье использованы как научные образцы, так и результаты людей, тестировавшихся частным образом в коммерческих лабораториях (компании 23andMe и FTDNA). Анализ производился по широкогеномным данным об аутосомных маркерах. Использовано несколько частично отличающихся панелей Illumina, некоторые образцы получены усечением полногеномных данных до широкогеномных. Для IBD-анализа применено усечение всех геномов до набора из 244 тысяч снипов, которые присутствуют во всех панелях. Для Admixture-анализа – усечение до 118 тысяч снипов, поскольку при этом виде анализа дополнительно исключаются близкорасположенные, «сцепленные» снипы. Сформированные выборки показаны на карте черными квадратами.

Две из них оказались за пределами карты – это потомки русских старожилов Сибири (5 человек) и Забайкалья (3 человека). Как иногда говорят, «чалдоны», или «челдоны». Цифры рядом с названием показывают размер каждой выборки. В случае указания размера через косую черту, число слева означает количество использованных образцов для выведения усреднений по компонентам Admixture, число справа – количество образцов, использованных для подсчета общих сегментов (имеются в виду малые выборки, которые сравниваются с большими. Они расположены под графиком сумм общих сегментов). Размеры больших выборок справа от IBD-графика (то есть тех, с которыми сравниваются малые выборки) следующие:
Balt 20 (литовцы, латыши и один схожий с ними геном с российско-латвийского пограничья);
Finnish-East 18 (восточные финны);
Karelian 18 (карелы);
Komi 19 (коми);
Mansi 9 (манси);
Mari 16 (марийцы);
Russian-Pomor 12 (русские поморы);
Saami 13 (саамы);
Udmurt 19 (удмурты);
Veps 14 (вепсы).
Выборки, использованные для построения карт, подробно не описываются, поскольку их очень много, а сами карты приводятся исключительно в иллюстративных целях.
Источник основной части научных образцов – Эстонский Биоцентр (геномы выкладываются здесь). Сложно перечислить, в какой из статей был впервые использован каждый геном, при желании эту информацию можно найти на сайте биоцентра. Кроме того, использованы выборки из проекта «1000 геномов», а также статьи Hellenthal et al, 2014 . Несмотря на небольшое количество геномов, очень полезными для анализа северного генофонда оказались полные сиквенсы из работы Wong et al, 2015
«Изюминкой» моих работ можно посчитать включение «коммерческих» геномов. К сожалению, кроме плюсов (добавление результатов популяций, не охваченных научным тестированием), у этого решения есть и минусы. Главный из них – автор не может доказать, что используемые образцы соответствуют строгим научным критериям формирования выборок. Геномы высылались мне для анализа энтузиастами генетической генеалогии, собравшимися на площадке forum.molgen.org , при условии, что файлы не будут передаваться третьим лицам. В выборку включались люди, все ближайшие предки (хотя бы 2-3 поколения – одиночный предок далее прадедушек и прабабушек уже влияет на результаты очень мало) которых происходили из нужной местности и относились к нужному этносу. За время существования проекта было обработано более 500 «коммерческих» геномов восточноевропейцев, однако большинство из них нельзя включить в «опорные» выборки из-за несоответствия указанному критерию либо из-за близкородственности с уже обработанными геномами. Тем не менее, для проверки полученных результатов они вполне пригодны. Таким образом, общий объем восточноевропейской выборки весьма велик.
Лично я уверен в добросовестности генеалогов-молгеновцев. Свое происхождение они нередко знают на столетия назад, поэтому с моей точки зрения «коммерческие» выборки временами могут являться даже более качественными, чем научные. Косвенным доказательством их корректности служит то, что полученные результаты без каких-либо противоречий вписываются в единую систему с научными выборками. При появлении нужных научных выборок «коммерческие» результаты будут либо окончательно подтверждены, либо исправлены и дополнены.
Автор выражает особую благодарность за помощь при формировании северных выборок Анатолию Воронцову, Владимиру Волкову и Владимиру Беданову. Благодарность выражается и всем участникам проекта.
Причина разницы в количестве образцов, использованных для IBD- и Admixture- анализа заключается в том, что часть научных геномов уже была использована при создании 27-компонентной модели. Поэтому получаемые для них результаты отличаются от всех остальных, не использовавшихся при выведении компонентов Admixture и исключены из сравнения (так называемый «эффект калькулятора»). Для IBD-анализа подобной проблемы не существует.
Одним из первых толчков к написанию данной статьи следует признать работу Андрея Хрунина и др. от 2013 года, где, насколько мне известно, впервые в научном сообществе был выделен «северо-восточный» европейский аутосомный полюс. Надо упомянуть, что любителями, например, уже упоминавшимся В.Вереничем, при анализе Admixture этот полюс выделялся и ранее. Позднее авторы и далее развивали тему изучения северного генофонда, в том числе выступив соавторами в упомянутой выше статье Wong et al.
Более широкую известность получила статья 2011 года “Генофонд Русского Севера: Славяне? Финны? Палеоевропейцы?” Елены Балановской и др, однако в ней исследовались однородительские маркеры, и аутосомы остались почти незатронутыми (кроме одиночного маркера CCR5del32).
Сходства и различия изученных русских выборок между собой и с соседними славянскими, балтскими, финскими народами подробно изучались в известной статье о генофонде славян и балтов (Алёна Кушняревич и др., 2015), подытоживающей накопленную на тот момент научным сообществом информацию, а также вышедшей в конце того же года монографии Олега Балановского «Генофонд Европы» , дающей подробный обзор европейского генофонда. В обеих работах проводился анализ по всем трем основным системам генетических маркеров (мужские и женские однородительские линии, и аутосомы).
Перечислить все имеющие отношение к анализу северного генофонда научные работы крайне сложно, и все равно остается вероятность пропустить что-то важное. Поэтому не буду дополнительно углубляться в тему, а перейду к следующей части.
Основные компоненты аутосомного генофонда северян.
При анализе северного генофонда можно выделить три его главных источника. Здесь я не останавливаюсь подробно на их выведении, ограничившись общим описанием и несколькими иллюстрациям, поскольку эти компоненты можно увидеть во многих научных работах.
Первый, наиболее древний слой – это «палеоевропейский» компонент, и поныне преобладающий у всех северных европейцев. Благодаря произведенной за последние годы расшифровке заметного количества геномов древних охотников-собирателей мы можем уверенно судить, что его корни тянутся на десятки тысячелетий в прошлое. Правда, в основном эти геномы относятся к более южным регионам Европы – от Испании (la Brana) до Воронежской области (Костёнки). Однако есть и результат «северянина» — это образец I0061 (из работы Haak et al. 2015 года) с Южного Оленьего острова, Карелия. Его возраст, согласно радиоуглеродной датировке, составляет около 7-7.5 тысяч лет. При его сравнении с современными выборками первая десятка с наибольшей долей совпадающих снипов (одиночных мутаций), по моим расчетам, оказалась следующей:
Эстонцы
Латыши и литовцы (объединенная «балтская» выборка)
Русские Каргополя («вологодская» выборка HGDP)
Поляки
Восточные финны
Карелы
Вепсы
Саами
Словаки
Западные финны
На момент сравнения у меня еще не была сформирована выборка беломорских русских (поморы) – не сомневаюсь, что она тоже заняла бы высокое место в этом «рейтинге палеоевропейскости».
Несмотря на географию, я не считаю «оленеостровца» представителем основной части предков современных прибалтийских финнов или собственно балтов — на эту роль лучше подходят более южные образцы. Думаю, что он ближе к предкам удмуртов (в рейтинге они отдалились из-за наличия у современных удмуртов восточноазиатского и степного влияния). Также любопытна его взаимосвязь с западнославянскими выборками. Вероятно, картина прояснится при появлении новых северных палеообразцов, а пока данных слишком мало, чтобы уверенно их структурировать.
Второй компонент можно назвать «уральско-сибирским». Его влияние заметно ниже, при этом в целом падает при продвижении с востока на запад. Видимо, изначальное происхождение компонента связано со смешением восточных, «сибирских» вариантов континуума древних охотников-собирателей Северной Евразии (западным вариантом которых являются палеоевропейцы) и пришельцев с юго-востока, принесших с собой восточноазиатские генетические варианты. Напрашивается предположение о взаимосвязи компонента с распространением в северо-восточной Европе языков уральской семьи, а также многих ветвей Y-гаплогруппы N. Согласно реконструкции Владимира Напольских, перед распадом прауральского языка его носители проживали в темнохвойной тайге западносибирского типа, вероятно, также частично захватывая Урал и Восточную Сибирь (см. «Предыстория уральских народов», 2001 ). Можно предположить, что при продвижении на запад «уральские» мужчины женились на местных женщинах, и в следующих поколениях доля «восточных» аутосомных вариантов снижалась, замещаясь «западными». В результате наблюдаемая картина при использовании разных систем генетических маркеров резко различается – если по Y-гаплогруппам влияние пришельцев весьма высоко, а кое-где преобладает подавляюще, то по аутосомным и мито-маркерам оно или совсем невелико, или находится в меньшинстве.
Среди современных европейских народов наиболее сильно этот аутосомный компонент проявляется у марийцев. Однако и у них он смешан с «палеоевропейским», поэтому для его выделения удобнее использовать результаты угорских народов Западной Сибири.
.")
Наконец, третий, наиболее поздний компонент – это вклад пришедших с юга восточнославянских переселенцев. Этот компонент также имеет палеоевропейскую основу (что затрудняет дифференцирование с первым компонентом), однако с некоторой добавкой «южных» влияний (предположительно, неолитических земледельцев Европы и степных групп бронзового либо железного века). Сложно оценить, насколько велико влияние восточных славян на финские и пермские народы. Некоторые следы этого можно увидеть, но какая его часть связана с русскими миграциями Средневековья и нового времени, а какая — с более ранними движениями населения (например, знаменитыми «шнуровиками», или контактами прафинноугров со степными индоевропейцами)? Точную оценку станет возможно сделать только после прочтения северных геномов дославянского периода, а пока что мы можем опираться лишь на результаты наших современников. Поэтому я принял в качестве рабочего предположения, что до XX века люди смешанного происхождения обычно либо входили в состав русских, либо это делали их потомки. Тогда влиянием русских на генофонд сохранившихся финнов и пермян можно условно пренебречь, а найденные у них «южные» аутосомные компоненты отнести на иные контакты.
Дальнейший анализ будет производиться в два этапа. На первом я попытаюсь раскрыть взаимосвязи между неславянскими народами Севера. На втором – опираясь на уже полученные результаты, провести анализ генофонда северных русских. Единственным исключением станет восточная половина выборки поморов – русские Пинеги и Мезени. Согласно реконструкции, в основной части они потомки дославянского населения Севера, с минимальным влиянием центральных и южных русских. Поэтому их результаты представляют большую ценность для понимания северного генофонда и включены уже в первый этап анализа. Разумеется, это не делает пинежан и мезенцев какими-то «неправильными» или «поддельными» русскими, как иногда воспринимают мои слова.
Этап 1. Анализ взаимосвязей между неславянскими народами Севера.

Поскольку IBD-анализ чувствителен к размеру выборок, в поморскую выборку (Russian-Pomor), кроме мезенцев и пинежан, для надежности добавлены близкие к ним генетически русские из низовьев Северной Двины. Komi-Zyryan-NE – это объединение результатов двух ижемских коми м одного близкого к ним коми из другой научной выборки, точное происхождение которого неизвестно. Komi-Zyryan-SW – объединение результатов двух прилузских коми, одного коми из Сыктывкара и двух близких к ним коми из научной выборки, точное происхождение которых неизвестно.
Как видите, количество общих сегментов хорошо отображает географию (проживающие рядом народы имеют больше возможностей генетически повлиять друг на друга). Например, у карел очень много общих сегментов с вепсами. Однако при этом у прибалтийских финнов больше общих сегментов с карелами, чем с вепсами, а у поморов и коми – чуть больше с вепсами, чем с карелами, но в целом близко. Таким образом, можно сказать, что с точки зрения аутосомного генофонда карелы – это смешение вепсов и восточных финнов.
По пикам сумм IBD-сегментов в общей «палеоевропейской» массе можно выделить отдельные «генетические полюса». Причина их появления – генетический дрейф в результате изоляции. Население Севера долгое время было очень редким, поэтому выделение полюсов происходило быстрее их размывания в результате смешения с соседями. Южнее в большинстве случаев этого не происходило, размывание шло быстрее выделения. Однако, к примеру, евреи-ашкенази за примерно тысячелетие генетически изолированного от окружающих существования сформировали хорошо выраженный полюс. Также хорошо формируют «генетические полюса» народы Северного Кавказа, благодаря географической изоляции (горный рельеф).
Чем более сильным оказался дрейф, тем выше будет пик на IBD-графике у соответствующего полюса. Возможно и формирование вторичных полюсов – когда смешиваются уже хорошо отдрейфовавшие популяции, а получившаяся группа позже испытывает дополнительный дрейф (например, так предположительно произошло с вепсами и карелами).
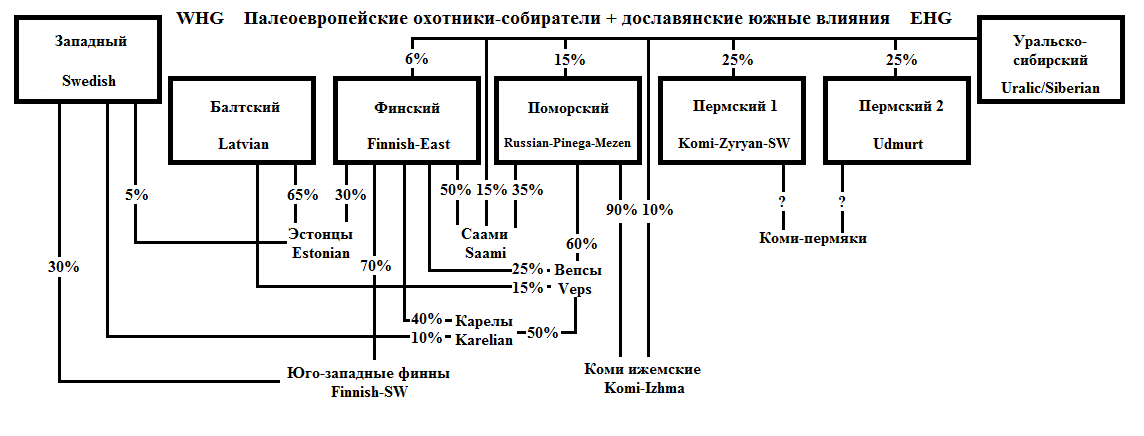
Основные генетические полюса, проявляющиеся на Севере, изображены на схеме внизу в черных прямоугольниках от «Балтский» до «Пермский 2». В верхней строке приведено условное название полюса, в нижней – выборка, для которой его влияние проявляется наиболее ярко. При вычислении влияния полюса на соседние популяции значения для этих выборок условно приняты за 100%. Выделение полюсов основано на анализе IBD-сегментов, а вероятные пропорции смешения получены сравнением компонентов Admixture (диаграмма с их значениями будет приведена далее).

В правом верхнем углу изображен гипотетический исходный «уральско-сибирский» компонент и его влияние на северные популяции (при оценке его влияния я в основном ориентировался на результаты выборки манси). В левом верхнем углу – «западный» компонент, проявляющийся на графиках Admixture у выборок, расположенных рядом с Финским заливом. Здесь за основу взяты результаты шведов, поскольку наиболее хорошо он выделяется у юго-западных финнов, а влияние шведов на их генофонд согласуется с известными из истории фактами. Впрочем, использование как манси, так и шведов – условность для целей моделирования. Это не значит, что соответствующее влияние шло именно от предков этих народов.
Центральные и южные русские (а также большинство остальных славян) тяготеют к «балтскому» полюсу.
Первые четыре полюса близкородственны между собой, в то время, как Пермский 2 (предки удмуртов) находится несколько поодаль, повлияв лишь на первый пермский полюс (поток генов от удмуртов к коми). Положение коми-пермяков точно неизвестно, но логично предположить, что они должны быть смесью влияний двух пермских полюсов (это предположение будет использовано при анализе результатов пермских русских).
Любопытно, что выборка северо-восточных (ижемских) коми проявила гораздо больше общего с поморами, чем с другими пермянами. Думаю, что это разделенные части единой древней популяции (назовем ее «чудь заволочская»). Большинство ее представителей вошло в состав русского народа, но крайняя северо-восточная часть перешла на пермский язык (либо изначально была не финно-, а пермскоязычной) и стала коми-ижемцами.
Довольно сложным получилось происхождение кольских саами – хотя в наибольшей степени они связаны с «финским» полюсом, сильно проявлены как «поморский» полюс, так и «уральско-сибирский» компонент, который тоже, вероятно, можно разбить на несколько полюсов. Как мне кажется, «уральско-сибирское» влияние у саами больше марийского, чем мансийского или ненецкого типа, но тут тяжело надежно выявить подробности. Кроме того, моделирование для саами произведено без участия Admixture, поэтому надежность реконструкции снижена.
И все же, насколько оправдано использовать результаты выборки восточных поморов в качестве 100% представителей дославянского генофонда? Результаты Admixture показывают, что они почти в точности вписываются между своими соседями с юго-запада (вепсы) и востока (коми-ижемцы).
.")

Как будет показано далее, вклинивающиеся между ними русские Каргополя аутосомно схожи с более южными и восточными выборками, такими, как русские Сольвычегодска. Восточные финны здесь отличаются настолько сильно из-за того, что для них выделен отдельный компонент Admixture (на диаграмме выше голубой) и отклонение по нему наиболее велико.
Могут выдвинуть возражение, что это русские настолько повлияли на генофонд коми и вепсов, что они стали походить на поморов. Но если уж русский колонизационный поток был настолько мощным, чтобы кардинально менять генофонд местного населения, то собственно русские выборки Севера в таком случае должны быть неотличимы от центральных и южных русских. А подобного не наблюдается.
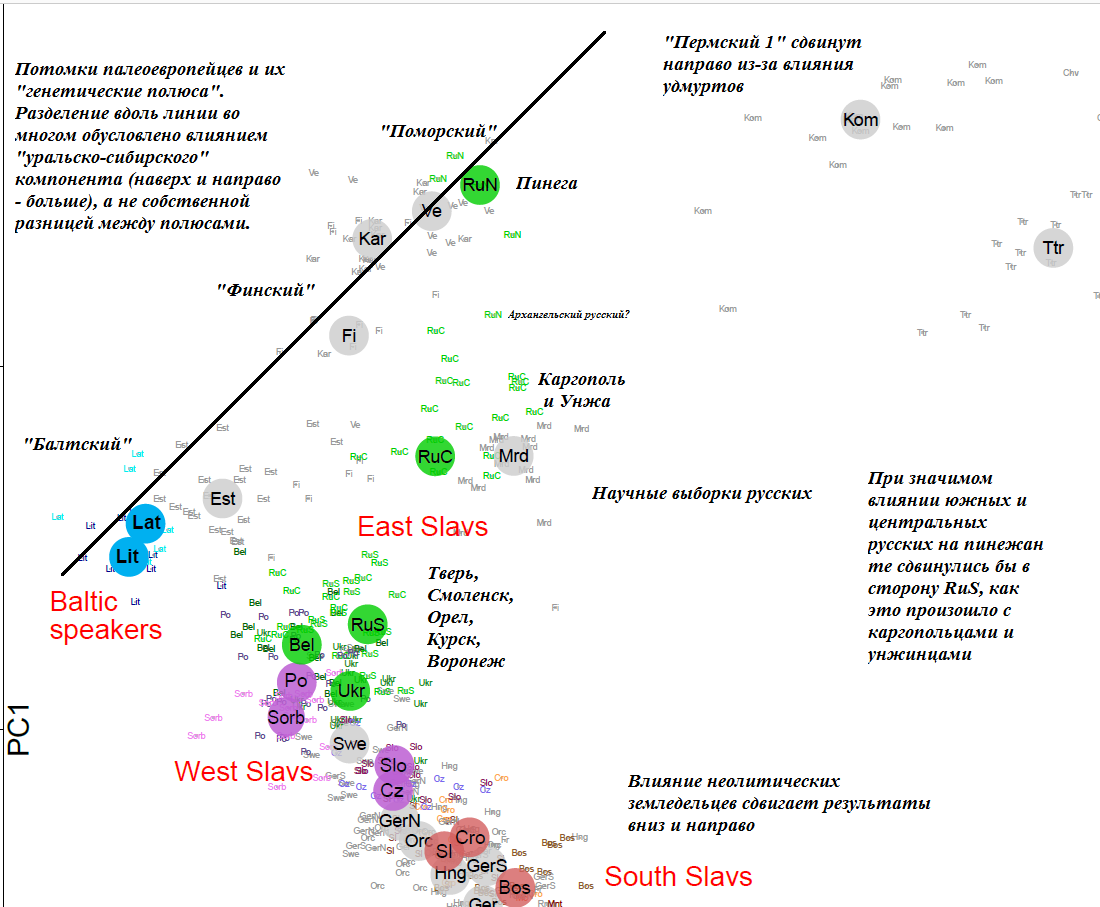
В качестве подкрепления выводов приведу часть графика главных (первая и вторая) компонент из упоминавшейся выше этапной работы Кушняревич и др., 2015 о генофонде славян и балтов. Здесь использованы только научные выборки. Черным курсивом даны мои комментарии и пояснения, мной же добавлена и черная линия, соединяющая наиболее «аутосомно северные» выборки:

Этап 2. Структура генофонда северных русских.
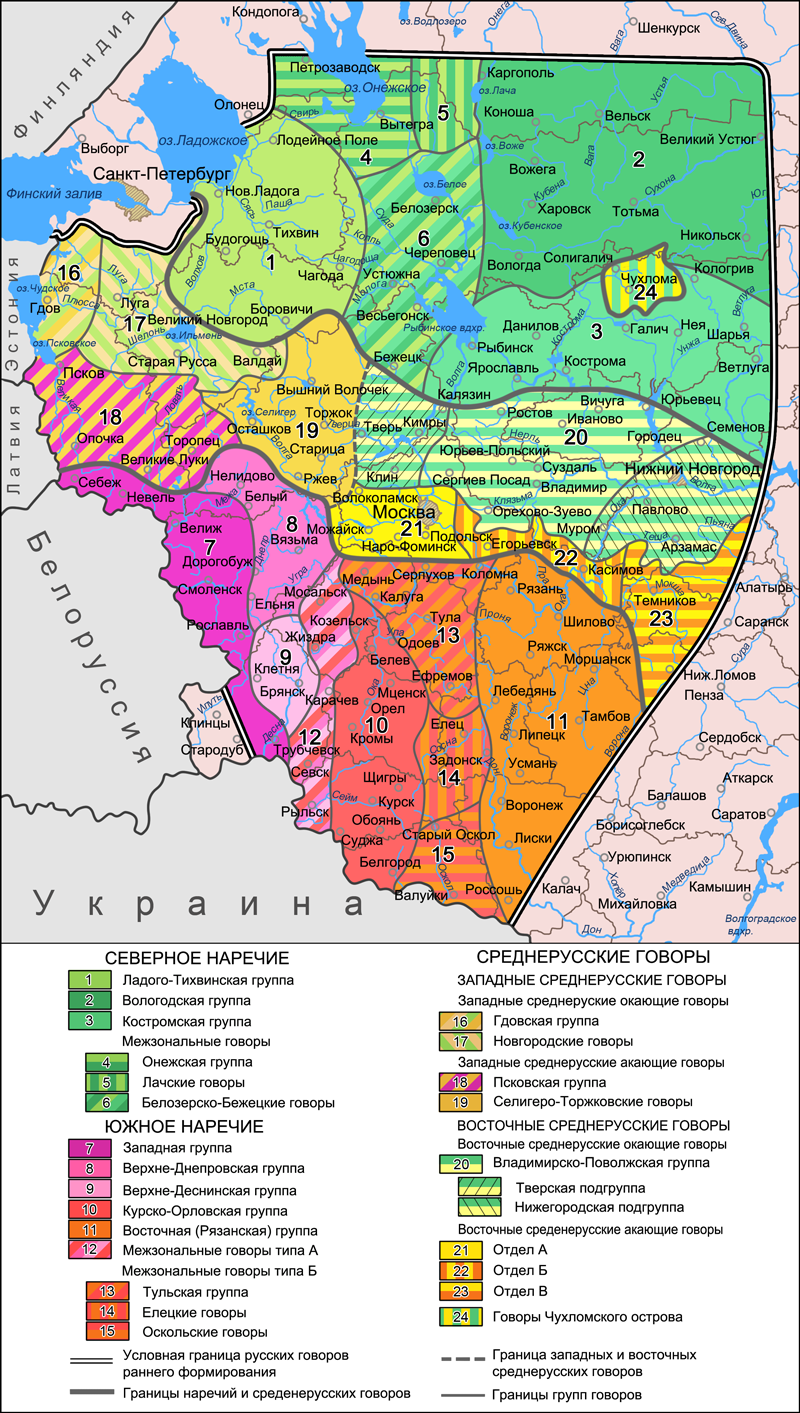
При решении вопроса, какие выборки отнести к северным русским, за основу взята карта русских говоров, составленная советскими лингвистами (по Русская диалектология / под ред. Р. И. Аванесова и В. Г. Орловой. М.: Наука, 1965). Она хорошо коррелирует с аутосомными данными, включая один спорный момент – жителей зон 16 и 17 (историческая Новгородская Земля, «словене ильменские») по некоторым признакам тоже можно отнести к северянам. Здесь это названо «западные окающие говоры». Как мы увидим, разница с зоной «западных акающих говоров» (Псковщина, «кривичи псковские») хорошо проявляется и в аутосомном генофонде. К сожалению, образцы из «чухломского острова» (зона 24) отсутствуют – было бы интересно сравнить их с соседями.

Кроме северян, использованы выборки новгородских русских (она должна представлять «новгородский» поток колонизации), псковская (для сравнения с новгородцами) и «ростовская» (она представляет не окрестности города Ростова, хотя есть и образец оттуда, но «ростовскую землю» и «низовский» поток колонизации в целом – использованы образцы от Углича до Иваново). Для отображения возможных колонизационных потоков «из глубин славянского мира» взята выборка смоленских русских. Из географических соображений на график добавлены тверские русские (без северо-восточной части Тверской области, относящейся уже к зоне северных говоров), и небольшая выборка муромчан. Сформировать выборку русских Владимирского Ополья не удалось – пришлось ограничиться «ростовцами».
Северяне принимали активное участие в процессе освоения русскими Урала и Сибири (долгое время основные пути туда проходили через Север). Поэтому в анализ включены выборки потомков русских старожилов Пермского края, южной части Западной Сибири и Забайкалья.

Отличить северян оказалось очень легко – если для русской выборки красная линия (пересечения с поморами) получилась выше или наравне с зеленой (пересечения с балтами) – перед нами северные русские. Если ниже – центральные, северо-западные, либо не показанные здесь южные.
Больше всего пересечений с балтами найдено у псковских русских. Псков, Смоленск и Тверь расположены на землях, которые когда-то населял славянский племенной союз кривичей. Археологи обнаруживают у них сильное балтское влияние и некоторые исследователи даже относят кривичей к ославяненым балтам. Впрочем, как я уже писал, большинство славян тяготеет к «балтскому» полюсу, и его можно с чистой совестью называть «балто-славянским». Просто у балтов он выражен несколько сильнее.
Очень своеобразны результаты новгородской выборки. У них относительно мало пересечений с поморами, но при этом проявлены пересечения с прибалтийско-финскими народами (это заметно при сравнении с соседями из Пскова и Твери). То есть новгородцы – северяне по параметру «есть предки из уральскоязычных народов», но не северяне по более узкому параметру «есть предки из чуди заволочской». Если вернуться к схеме взаимосвязей дославянского населения Севера, можно увидеть, что «уральско-сибирский» аутосомный компонент до Балтики практически не добрался, растворившись по дороге. На графике это отражается небольшим количеством пересечений с выборкой манси у новгородцев. По сути, здесь мы видим базовый уровень родства палеоевропейских времен (20-25 сМ). У карел и вепсов он повышается до 35, а у коми, удмуртов и саами – до 50-60 сМ. Для сравнения, у хантов этот показатель составил 229 сМ.
Для просчета возможных вариантов происхождения северян использован оракул на основе сравнения сумм IBD-сегментов в режиме «комбинация не более, чем пяти предковых популяций». В качестве опорных взяты выборки вепсов, карел, удмуртов, юго-западных коми-зырян («ижемскую» выборку включать нет смысла, поскольку она будет дублировать поморов с добавлением лишнего «уральско-сибирского» влияния), русских Пинеги-Мезени (поморы), Новгорода, Пскова, Ростова и Смоленска. Сравнение идет по суммам общих сегментов с представителями четырех «палеоевропейских» полюсов (балты, восточные финны, поморы, коми), а также четырьмя дополнительными выборками (вепсы, манси, саами, марийцы).
Для начала проверим оракул на новгородцах. Здесь и далее приводятся пять результатов с наименьшим среднеквадратичным отклонением (значение после @).
Russian-Smolensk 90% + Karelian 10% @ 7
Russian-Pskov 5% + Russian-Smolensk 85% + Karelian 10% @ 7,02
Vepsa 5% + Russian-Smolensk 85% + Karelian 10% @ 7,54
Vepsa 5% + Russian-Pskov 5% + Russian-Smolensk 85% + Karelian 5% @ 7,56
Russian-Pinega-Mezen 5% + Russian-Smolensk 85% + Karelian 10% @ 7,72
Russian-Pinega-Mezen 5% + Russian-Pskov 5% + Russian-Smolensk 80% + Karelian 10% @ 7,85
Лучше всего новгородцы моделируются, как смесь смоленских русских и небольшой доли карел или вепсов. Результат выглядит весьма правдоподобно. Хотя, скорее всего, источником «финского» влияния выступали не столько карелы, сколько более близкие географически народы (летописные чудь, водь и т.д.). Если вновь вернуться к схеме взаимоотношений дославянского населения из начала статьи, то результаты эстонцев показывают – у местных финнов должно было быть меньше «финского» и больше «балтского» влияния. Таким образом, более вероятное соотношение пришлого и местного элемента у новгородцев не 90 на 10, а примерно 80 на 20.
Перейдем к северянам. Как и на графике, выборки расположены в порядке убывания сумм IBD-пересечений с поморами – от более выраженных северян к менее выраженным.
Важная ремарка – я попытался просчитать не только соотношение вклада местного населения и пришельцев-славян, но и соотношение вклада различных групп этих пришельцев. Однако разница между ними относительно невелика, поэтому надежность дополнительного разбиения заметно ниже. Пожалуйста, имейте это в виду при анализе результатов.
Поморы с низовьев Северной Двины:
Russian-Pinega-Mezen 75% + Russian-Pskov 5% + Russian-Novgorod 15% + Karelian 5% @ 4,89
Russian-Pinega-Mezen 75% + Russian-Novgorod 20% + Karelian 5% @ 4,93
Vepsa 5% + Russian-Pinega-Mezen 70% + Russian-Pskov 15% + Russian-Novgorod 5% + Karelian 5% @ 5,01
Russian-Pinega-Mezen 70% + Russian-Pskov 20% + Karelian 10% @ 5,03
Russian-Pinega-Mezen 80% + Russian-Pskov 5% + Russian-Novgorod 5% + Russian-Smolensk 5% + Karelian 5% @ 5,08
Vepsa 5% + Russian-Pinega-Mezen 70% + Russian-Pskov 20% + Karelian 5% @ 5,09
Мы уже знали, что они весьма близки пинежско-мезенской выборке, поэтому затруднений у оракула не возникло. Доминирует местный элемент, плюс показывается некоторое влияние новгородцев и карел. В некоторых комбинациях алгоритм «расщепляет» влияние новгородцев на составляющие, которые мы уже видели в предыдущем примере (русские Пскова или Смоленска плюс карелы или вепсы).
Русские Сольвычегодска:
Russian-Pinega-Mezen 35% + Russian-Rostov 30% + Russian-Novgorod 15% + Komi-Zyryan-SW 10% + Karelian 10% @ 3,12
Russian-Pinega-Mezen 40% + Russian-Novgorod 40% + Komi-Zyryan-SW 10% + Karelian 5% + Udmurt 5% @ 3,65
Russian-Pinega-Mezen 35% + Russian-Pskov 10% + Russian-Rostov 35% + Komi-Zyryan-SW 10% + Karelian 10% @ 3,66
Russian-Pinega-Mezen 35% + Russian-Rostov 35% + Russian-Smolensk 10% + Komi-Zyryan-SW 10% + Karelian 10% @ 3,69
Russian-Pinega-Mezen 40% + Russian-Pskov 5% + Russian-Novgorod 35% + Komi-Zyryan-SW 15% + Karelian 5% @ 3,73
Здесь с автохтонной стороны к «поморскому» компоненту добавляется влияние коми, что выглядит логично с точки зрения географии. Со славянской же стороны алгоритм подставляет то ростовцев, то новгородцев. Это тоже можно посчитать логичным – Сольвычегодск расположен рядом с важнейшим перекрестком северных речных путей, где реки Сухона, Юг и Вычегда превращаются в Двину. Здесь долгое время шла борьба между «низовцами» и новгородцами (опорным пунктом первых был расположенный чуть западнее Великий Устюг). Как известно, соперничество Новгорода и Ростовской земли/Владимирской Руси/Великого Княжества Московского в конечном итоге закончилось поглощением Новгорода и его бывших северных владений Москвой.
Однако я не исключаю и варианта, описанного в ремарке – алгоритм просто не справился с разделением влияния групп славян. Само же соотношение пришлого и местного элемента в генофонде сольвычегодцев можно оценить, как 50 на 50.
.")
Русские Пермского края:
Russian-Pinega-Mezen 40% + Russian-Pskov 15% + Russian-Novgorod 15% + Komi-Zyryan-SW 15% + Udmurt 15% @ 3,49
Russian-Pinega-Mezen 40% + Russian-Novgorod 20% + Russian-Smolensk 10% + Komi-Zyryan-SW 15% + Udmurt 15% @ 3,78
Russian-Pinega-Mezen 40% + Russian-Pskov 25% + Russian-Smolensk 5% + Komi-Zyryan-SW 15% + Udmurt 15% @ 3,8
Russian-Pinega-Mezen 40% + Russian-Pskov 30% + Komi-Zyryan-SW 15% + Udmurt 15% @ 3,85
Russian-Pinega-Mezen 40% + Russian-Pskov 25% + Russian-Rostov 5% + Komi-Zyryan-SW 15% + Udmurt 15% @ 3,95
Первоначальное заселение края русскими происходило северным путем, через Вычегду и волоки в бассейн Камы. Путь по Волге и низовьям Камы был перекрыт казанцами. Судя по результатам, Пермь стала областью вторичной колонизации, куда в основном переселялись уже северные русские – иначе не объяснить столь большую долю «поморского» компонента. Видимо, комбинации «40% поморы + 30% северо-западные русские» отражают вклад северян (с вероятным добавлением небольшой доли других групп русских) поскольку такая смесь примерно соответствует русским Сольвычегодска, которых я условно принял за типичных представителей северян. Тогда «15% коми-зыряне + 15% удмурты» — это преимущественно отображение вклада местного пермского населения. К сожалению, результатов коми-пермяков у меня нет, но с точки зрения географии они примерно так и должны выглядеть. На графике хорошо заметно повышение у пермских русских количества общих сегментов как с коми-зырянами (по этому показателю они на первом месте среди всех русских выборок), так и с удмуртами (второе место после одной специфичной вятской выборки). Однако здесь вклад дославянского населения в генофонд местных русских следует признать более низким, чем в предыдущих случаях – ведь «поморский» компонент на этот момент уже необходимо считать русским. Если взять в качестве образца переселенцев-северян русских Сольвычегодска, то соотношение пришлого и местного элемента в генофонде пермских русских можно оценить, как 75 на 25, или даже 80 на 20 (поскольку “пермский” компонент частично присутствует уже у сольвычегодцев).
Русские Каргополя:
Russian-Pinega-Mezen 35% + Russian-Pskov 20% + Russian-Novgorod 30% + Komi-Zyryan-SW 5% + Udmurt 10% @ 1,67
Vepsa 5% + Russian-Pinega-Mezen 30% + Russian-Pskov 25% + Russian-Novgorod 25% + Udmurt 15% @ 1,83
Russian-Pinega-Mezen 35% + Russian-Rostov 15% + Russian-Smolensk 35% + Karelian 5% + Udmurt 10% @ 1,84
Russian-Pinega-Mezen 35% + Russian-Pskov 10% + Russian-Rostov 20% + Russian-Novgorod 25% + Udmurt 10% @ 1,95
Russian-Pinega-Mezen 35% + Russian-Rostov 30% + Russian-Novgorod 25% + Komi-Zyryan-SW 5% + Udmurt 5% @ 1,98
Знаменитая выборка из Human Genome Diversity Project, долгое время представлявшая в большинстве научных работ всех русских, что вызывало у многих бурное негодование (в том числе временами и у меня). Каргополь расположен рядом с границами Карелии, и само его название выводят от карельского «медвежья сторона». Поэтому я ожидал найти здесь немалое влияние карел. Однако ничего подобного не наблюдается — результаты схожи с полученными для более восточных выборок северных русских (сольвычегодцы). Откуда там взялись пересечения с удмуртами, непонятно. Впрочем, их не так и много – вероятно, удмурты введены алгоритмом в попытке сбалансировать комбинации, а реального влияния почти нет.
Возможно, местные финны оказались более схожи по аутосомному портрету не с используемой мной карельской выборкой из Приладожья, а с поморами. Еще один приходящий в голову вариант объяснения – каргопольская выборка представляет позднейших русских переселенцев с юга и востока (Сухона, Вычегда и т.д.). Город стоял на торговом пути, идущем от Вологды и Белозерска, то есть из сферы влияния «низовцев». Поэтому взаимосвязь тут возможна. На карте диалектов Каргополь размещен у границы вологодских и межзональных (переходных к ладого-тихвинским) говоров, то есть тяготеет к Сухоне.
Из-за подобной неопределенности оценка вклада местного и пришлого населения не производится.
Русские Устюжны:
Russian-Pinega-Mezen 15% + Russian-Rostov 30% + Russian-Novgorod 45% + Karelian 5% + Udmurt 5% @ 9,22
Russian-Pinega-Mezen 25% + Russian-Rostov 20% + Russian-Novgorod 50% + Udmurt 5% @ 9,24
Vepsa 5% + Russian-Pinega-Mezen 15% + Russian-Rostov 30% + Russian-Novgorod 45% + Udmurt 5% @ 9,24
Russian-Pinega-Mezen 25% + Russian-Rostov 15% + Russian-Novgorod 50% + Russian-Smolensk 5% + Udmurt 5% @ 9,26
Russian-Pinega-Mezen 25% + Russian-Pskov 5% + Russian-Rostov 15% + Russian-Novgorod 50% + Udmurt 5% @ 9,33
Согласно В. В. Седову, культура сопок, характеризующая ильменских словен, распространялась до бассейна реки Мологи, где и расположена Устюжна (Седов В. В. Древнерусская народность. Историко-археологическое исследование. М., 1999). Таким образом, район Устюжны начал заселяться будущими новгородцами очень рано. Неудивительно, что здесь преобладает «славянское» влияние, причем больше новгородского типа. На сдвиг в «новгородскую» сторону также могла оказать влияние проживавшая рядом летописная весь или (в более позднюю эпоху) тверские карелы.
Надо учесть, что устюжнинская выборка очень мала – всего два человека. В большинстве случаев такие выборки мной не используются, однако этот район хотелось тоже охватить анализом. В результате отклонение даже для самой лучшей комбинации довольно велико – 9.22, что говорит об усилении погрешности. Хотя сами по себе результаты выглядят нормально (Udmurt 5% можно спокойно пренебречь, это не 10-15%, как у каргопольцев).
Русские Вятки:
С размером выборки вятских русских проблем нет, однако она оказалась слишком неоднородной. Поэтому я вывел из нее результаты двух вятчан с сильным коми-пермяцким или удмуртским влиянием, которые превратились в отдельную выборку северо-восточных вятских русских (Russian-Vyatka-NE). На графике они показаны отдельно, однако приводить для них оракул я не вижу смысла. Для основной же выборки получены следующие варианты:
Vepsa 15% + Russian-Pinega-Mezen 5% + Russian-Smolensk 60% + Udmurt 20% @ 4,86
Vepsa 15% + Russian-Pinega-Mezen 5% + Russian-Pskov 5% + Russian-Smolensk 55% + Udmurt 20% @ 4,89
Vepsa 10% + Russian-Pinega-Mezen 10% + Russian-Novgorod 5% + Russian-Smolensk 55% + Udmurt 20% @ 4,99
Vepsa 10% + Russian-Pinega-Mezen 10% + Russian-Rostov 5% + Russian-Smolensk 60% + Udmurt 15% @ 5,08
Russian-Pinega-Mezen 25% + Russian-Smolensk 60% + Udmurt 15% @ 5,2
Согласно уже упоминавшейся формуле, комбинацию «русские Смоленска плюс немного вепсов» можно интерпретировать, как новгородцев (просто для алгоритма оказалось чуть удобнее разложить их таким образом). И тогда вятские русские получаются потомками новгородцев с некоторым местным влиянием (меньше, чем у пермских русских). В первом приближении интерпретация выглядит нормально, однако есть сомнения – почему тогда на графике провален характерный для новгородцев восточнофинский компонент, а поднят именно вепсский? Возможно, вепсы в комбинациях и обозначают (хотя бы частично) потомков летописной веси, влившихся в состав северян? Тогда для компенсации смоленские русские в комбинациях тоже должны частично обозначать переселенцев из коренной России.
Обе интерпретации видятся равноправными. Однако результаты русских Унжи навели меня на мысль о возможном третьем варианте, который остается чистым теоретизированием, но выглядит интересно. Об этом чуть ниже.
Русские Унжи:
Vepsa 5% + Russian-Pinega-Mezen 10% + Russian-Novgorod 70% + Komi-Zyryan-SW 10% + Udmurt 5% @ 1,75
Vepsa 5% + Russian-Pinega-Mezen 10% + Russian-Rostov 25% + Russian-Novgorod 50% + Komi-Zyryan-SW 10% @ 1,75
Vepsa 15% + Russian-Rostov 20% + Russian-Novgorod 50% + Komi-Zyryan-SW 5% + Udmurt 10% @ 1,76
Russian-Pinega-Mezen 15% + Russian-Novgorod 70% + Komi-Zyryan-SW 10% + Udmurt 5% @ 1,78
Russian-Pinega-Mezen 15% + Russian-Novgorod 50% + Russian-Smolensk 20% + Karelian 5% + Udmurt 10% @ 1,81
Выборка состоит из научных образцов Russian_Kostroma, плюс один геном из FTDNA (он не выбивается из общей тенденции). Результаты схожи с полученными для русских Вятки, за вычетом ослабления «пермского» компонента и усиления «прибалтийско-финского». По моему мнению, выглядят они странно, поскольку именно река Унжа была одной из дорог, по которой шла «низовская» колонизация Севера. Если где и должны в результатах преобладать Russian-Rostov, так это здесь.
В качестве возможного объяснения у меня родилась гипотеза, что под новгородцев в результатах северных русских может маскироваться летописная меря.
В наши дни этот народ не существует. Есть версия, что потомки мери – современные марийцы, однако мне она кажется неверной. И вот почему:
Если летописные чудь и пермь достоверно оставили след в русском генофонде, логично предполагать, что свой вклад должна была внести и меря. Однако марийцы формируют свой, хорошо выраженный «генетический полюс». Не заметить их влияние на генофонд русских было бы невозможно, но его следов нет. Аутосомные родственники марийцев известны – по моим исследованиям, в первую очередь это чуваши, и в меньшей степени – казанские татары.
Следовательно, чтобы вклад мери был трудноразличим, она должна быть генетически схожей с новоприбывшими в регион восточными славянами (аналогичное рассуждение справедливо и для двух других исчезнувших летописных племен – мещеры и муромы). Это вполне вероятно, поскольку известны результаты геномного анализа соседей мерян по региону, сохранивших языки уральской группы до наших дней – эрзян и мокшан. Их аутосомная основа – та же самая, что у балтов и славян («балтский генетический полюс»), лишь с некоторым «уральско-сибирским» налетом. У эрзян и мокшан нет «прибалтийско-финского акцента», однако северо-западными соседями мерян была летописная весь, современными потомками которой считаются вепсы. Следовательно, взаимосвязь с прибалтийскими финнами вполне возможна и даже вероятна. Вот и источник «вепсского» компонента у русских Унжи и Вятки.
А выборка, относящаяся к «балтскому полюсу», но с некоторым «прибалтийско-финским акцентом» — для оракула это и есть новгородцы.
С другой стороны, в противоречие с этой версией вступают результаты «ростовской» выборки русских – у них повышения количества пересечений с прибалтийско-финскими выборками не наблюдается (за исключением небольшого «бугорка» на графике пересечений с саами, которые очень специфические финны). А Ростов тоже относят к исторической территории мери. Впрочем, считается, что на костромщине меряне исчезли гораздо позже и имели больше возможностей оказать свое влияние. К тому же костромская меря могла заметно отличаться от ростовской.
Подытоживая, «мерянская» версия выступает здесь в качестве игры ума и не претендует на доказательность. Численную оценку влияния можно дать лишь очень грубо, исходя из компонентов Admixture. Если гипотетическая меря находилась в аутосомном смысле примерно посередине между современными вепсами и эрзянами, ее вклад в генофонд русских Унжи должен составлять менее половины (этого хватит для достижения нужного эффекта).
Кроме того, возможность влияния новгородцев эти рассуждения никак не отменяют. Соотношение «новгородского» и «мерянского» вкладов у северян может быть любым, поскольку структурно они схожи.
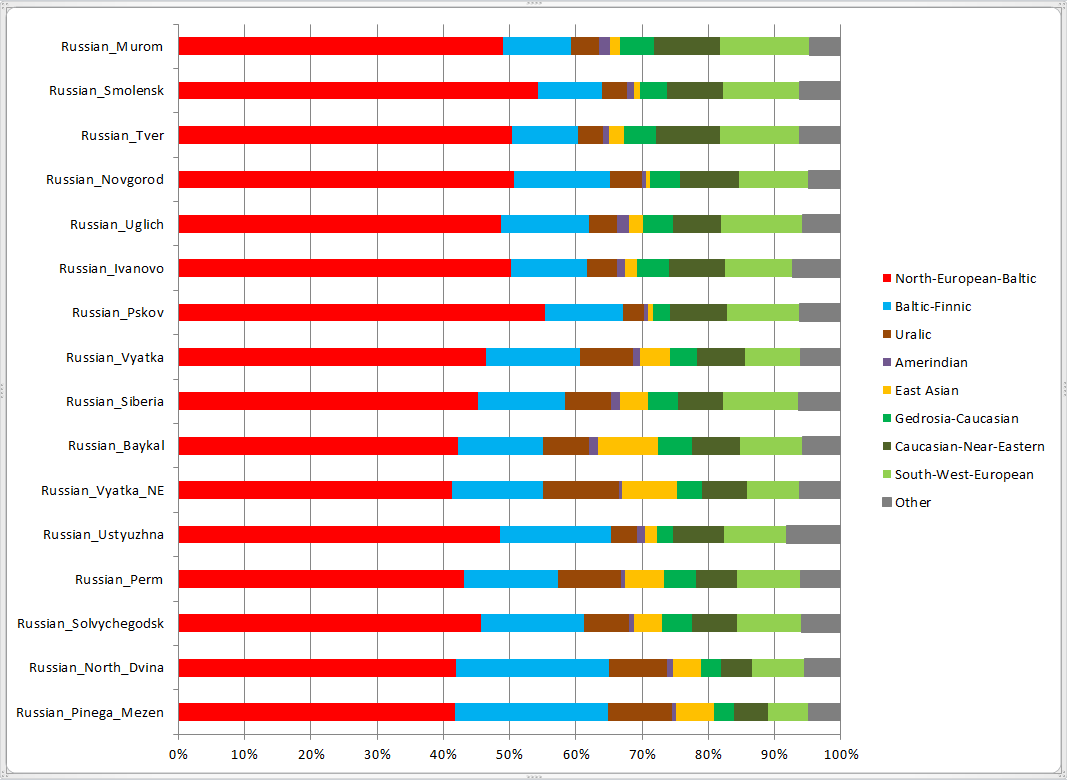
Для наглядности результаты IBD-оракула сведены в общую схему. Тверская и ростовская выборки получились близкими к смоленской и новгородской, с небольшим сдвигом в сторону поморского и пермского полюсов (их сектора занимают по 5% у тверской и по 10% у ростовской выборки). Для них используемая модель уже не вполне применима (вероятно, IBD-пересечения со многими полюсами могут восходить к более древним временам, чем у северных русских. Также возможны влияния других полюсов, неучтенных в модели.), однако для иллюстрации решено разместить и их результаты. Компоненты со значениями менее 5% не отображались.


Ростовская выборка здесь разбита на две части — Иваново и Углич. Каргопольская и унжинская выборки исключены полностью, поскольку участвовали в первоначальном выведении компонентов Admixture.
Читатель мог заметить, что при анализе результаты сибирских и забайкальских русских старожилов оказались пропущены. Дело в том, что применять для них ту же модель было бы некорректно – ведь на генофонд русских Сибири и Дальнего Востока могли заметно повлиять не только северные популяции, но и другие восточные славяне, а также народы Сибири. Поэтому была использована новая модель, оракул на основе сравнения пропорций компонентов Admixture в режиме «не более четырех предковых популяций» (расчет пяти предков при большом количестве выборок слишком затратен по времени). Опорные популяции должны примерно перекрывать основные возможные варианты (исключена лишь экзотика, наподобие пленных шведов, отправленных в Сибирь после Полтавы – в любом случае их возможный вклад слишком мал). Для этого использованы следующие выборки:
Altaian – алтайцы (3)
Bashkir_East – восточные башкиры (2)
Belarusian_Minsk – белорусы (центр) (5)
Buryat – буряты (3)
Evenk – эвенки (3)
Khanty – ханты (4)
Polish – поляки (15)
Russian_Bryansk – русские Брянска (6)
Russian_Don_Cossack – русские с Дона (2)
Russian_Ivanovo – русские Иваново (2)
Russian_North_Dvina – русские поморы (Двина) (5)
Russian_Novgorod – русские Новгорода (2)
Russian_Pskov – русские Пскова (4)
Russian_Ryazan – русские Рязани (3)
Russian_Smolensk – русские Смоленска (3)
Russian_Solvychegodsk – русские Сольвычегодска (6)
Russian_Ustyuzhna – русские Устюжны (2)
Russian_Ural_West – русские Перми и Екатеринбурга (7)
Russian_Vyatka – русские Вятки (4)
Ukrainian_Poltava – украинцы Полтавы (3)
Ukrainian_Slobozhanshtchina – восточные украинцы (3)
При использовании IBD-метода как забайкальцы, так и южносибирские «чалдоны» оказались северянами по критерию превышения суммы сегментов с поморами над суммой с балтами, хотя и менее выраженными, чем большая часть выборок северных русских (думаю, это говорит о том, что генофонд сибиряков сформирован не только ими). Посмотрим, что покажет Admixture.
Русские Забайкалья:
Evenk 5% + Russian_Don_Cossack 15% + Russian_Solvychegodsk 35% + Russian_Ural_West 45% @ 1,9
Bashkir_East 5% + Evenk 5% + Russian_Don_Cossack 15% + Russian_Solvychegodsk 75% @ 1,91
Evenk 5% + Khanty 5% + Russian_Don_Cossack 20% + Russian_Solvychegodsk 70% @ 1,99
Bashkir_East 5% + Evenk 5% + Russian_Bryansk 15% + Russian_Solvychegodsk 75% @ 1,99
Bashkir_East 5% + Evenk 5% + Russian_Solvychegodsk 75% + Ukrainian_Slobozhanshtchina 15% @ 2,03
В забайкальскую выборку включены потомки русских старожилов Забайкальского Края и один образец из северо-восточной части Иркутской области. В отличие от Севера, здесь смешение русских переселенцев с местным населением происходило в очень ограниченных масштабах. Видимо, различия оказались слишком велики. Оракул предполагает примерно 5% влияния народов Восточной Сибири, которых в модели представляют эвенки (на графике Admixture это проявляется, как повышение доли East Asian). Остальная часть распределяется между классическими северными русскими (Сольвычегодск), русскими Перми (в части комбинаций они показаны, как смесь русских Сольвычегодска и небольшого количества башкир либо хантов, которые здесь представляют уральскую сторону пермяков) и южными русскими либо украинцами. Это не значит, что среди предков забайкальцев не могло быть, к примеру, центральных русских – но в таком случае их вклад находился внутри диапазона между северянами и южанами и в модели разделился между этими крайними влияниями. Как и ожидалось, северный генофонд преобладает (80/15/5).
Русские южной части Западной Сибири:
Polish 15% + Russian_Solvychegodsk 20% + Russian_Ural_West 55% + Ukrainian_Poltava 10% @ 1,45
Polish 25% + Russian_North_Dvina 5% + Russian_Solvychegodsk 15% + Russian_Ural_West 55% @ 1,46
Polish 20% + Russian_North_Dvina 10% + Russian_Ural_West 60% + Ukrainian_Poltava 10% @ 1,46
Polish 15% + Russian_Ustyuzhna 10% + Russian_Ural_West 65% + Ukrainian_Poltava 10% @ 1,46
Polish 25% + Russian_North_Dvina 5% + Russian_Ustyuzhna 5% + Russian_Ural_West 65% @ 1,47
Сибирская выборка составлена из жителей Новосибирской, Кемеровской, Томской областей и Алтайского края. Здесь оракул не обнаруживает даже 5% вклада дославянского населения (возможно, небольшая его доля способна «прятаться» в завышении вклада уральцев). В остальном же результаты однотипны с полученными для забайкальских русских – смесь северного и южного генофонда с преобладанием северного (75/25). Поляки аутосомно очень схожи с украинцами и южными русскими, нередко до неотличимости, поэтому их наличие в комбинациях не удивляет. Не думаю, что их вклад в генофонд южносибирских старожилов достигает 15-25 процентов, хотя чем черт не шутит. Скорее комбинация с поляками просто оказалась чуть удобнее, чем с южными русскими или украинцами.

Сибирь очень велика и наверняка во многих местах ситуация развивалась по другим сценариям (в качестве примера достаточно вспомнить затундренных крестьян). И все же я думаю, что в большинстве случаев генофонд старожилов был схож с двумя исследованными здесь выборками. В наши же дни «среднего сибирского русского» навряд ли можно назвать выраженным северянином – массовое переселенческое движение конца XIX – начала XX века в Сибирь и более поздние события размыли сформировавшийся за три предыдущих столетия генофонд и должны были сделать сибиряков более схожими с центральными и южными русскими.
Общую долю северных русских среди великороссов на 1795 год можно оценить, как 20-25%, в зависимости от отнесения к северянам жителей северо-западных губерний. Оценка сделана мной, основываясь на подсчетах численности русского населения по регионам согласно работе: Кабузан В.М. «Народы России в XVIII в.: Численность и этнический состав». М., 1990. Таким образом, вклад северян в генофонд русского народа достаточно значим.
Карта схожести по пропорциям Admixture для русских Белого Моря уже была показана ранее. Для сравнения приведу и результаты из двух других углов «северного треугольника» карты для русских Новогорода и русских Перми.


Одна из основных сложностей при проведении аутосомного анализа заключается в том, что влияния всех эпох складываются в генофонде, наслаиваясь друг на друга. Чем дальше мы забираемся вглубь времён, тем больше позднейших наслоений следует учесть и попытаться убрать. Происхождение сибирских и уральских русских хорошо реконструируется, опираясь на результаты современных популяций. Полученные реконструкции соответствуют данным лингвистов и этнографов – действительно, мнение о том, что Урал первоначально заселялся русскими преимущественно с Севера, а Сибирь – преимущественно с Севера и Урала, но с заметным влиянием переселенцев из других регионов, можно считать практически общепринятым. Например (из Википедии):
«Сибирские старожильческие говоры, хотя и происходят генетически от северного наречия, утратили часть его архаических черт под влиянием говоров переселенцев с юга России. Вятские говоры и пермские говоры, размещённые на территории Кировской, Пермской и Свердловской областей считаются самыми архаичными среди всех говоров северного наречия, поскольку эти говоры лучше других сохраняют фонетику и морфологические особенности наречия Новгородской земли XIII—XIV веков.»
Из «наказа» тобольским воеводам (1596 год):
«служилых людей в пашню вваживать, чтобы себе пашню пахали и впред бы с Руси хлебных запасов посылати меньше прежнего, и велети пашенных и посадцких людей призывать из Перми, с Вятки, с Солей Вычеготцких на льготу Охочих людей»
При движении на шаг далее в прошлое, начинаются сложности. Насколько достоверно современные новгородские русские и русские Пинеги-Мезени могут представлять средневековых новгородцев и «чудь заволочскую»? Этого мы точно не знаем, хотя косвенные соображения говорят, что могут. Верна ли моя гипотеза о влиянии летописной мери на часть северных русских, или это просто игра ума? Нет нынче мерян, чтобы сравнить. И все же от той эпохи сохранилось достаточно много, чтобы строить детальные предположения.
Предшествующие же эпохи пока обрисовываются только очень крупными мазками. Здесь я не касаюсь результатов, полученных археологами и антропологами. Вероятно, в какой-то момент после «оленеостровца» в регион пришли люди с юга, предки будущих восточных славян, балтов и финнов. Возможно, они говорили на индоевропейских языках, возможно, нет («черепки не говорят»). В какой-то момент часть из них перешла на языки уральской семьи, и я предполагаю, что это связано с притоком «восточных» генетических вариантов. Затем некоторые из потомков перешедших стали индоевропейцами (вновь?), войдя в состав русского народа.
Поэтому очень важно получение большого количества расшифровок древних геномов, представляющих разные эпохи и разные регионы. «Сетка» результатов даст опору, позволяющую реконструировать происходившие миграции с высокой достоверностью. К счастью, в последние годы эта область науки бурно развивается, и я надеюсь, что скоро нас ожидает много новых, интересных результатов.
























.")

.")


.")





 и женского (розовые стрелки) населения в составе неолитической и степной миграций.")




{kind=link}
Для отправки комментария необходимо войти на сайт.