Согласно общепринятому определению, филогене́тика, или филогенети́ческая система́тика — область биологической систематики, которая занимается идентификацией и прояснением эволюционных взаимоотношений среди разных видов жизни на Земле, как современных, так и вымерших. Эволюционная теория утверждает, что сходство среди индивидуумов или видов часто указывает на общее происхождение или общего предка. Потому взаимоотношения, установленные филогенетической систематикой, часто описывают эволюционную историю видов и их филогенез, исторические взаимоотношения между ветвями организмов или их частей, например, их генов. Филогенетическая таксономия, являющаяся ответвлением, но не логическим продолжением филогенетической систематики, занимается классификацией групп организмов согласно степени их эволюционных отношений.
В понятийный аппарат ДНК-генеалогии филогенетика вошла в виде одного из своих направлений — кладистики. Характерные особенности кладистической практики состоят в использовании так называемого кладистического анализа (строгой схемы аргументации при реконструкции родственных отношений между таксонами), строгом понимании монофилии и требовании взаимно-однозначного соответствия между реконструированной филогенией и иерархической классификацией. Кладистический анализ — основа большинства принятых в настоящее время биологических классификаций, построенных с учётом родственных отношений между живыми организмами. Разумеется, кладистический анализ в ДНК-генеалогии не подразумевает анализ родственных отношений между таксонами-биологическими видами. Тем не менее, в силу схожести филогенетической схемы и привычной генеалогу схемы родственных отношений (генеалогического дерева), некоторые методы кладистики были взяты на вооружение и в ДНК-генеалогии. В-первую очередь, это касается филогений Y хромосомы и митохондриальной ДНК, восходящих к Y-хромосомному Адаму и митохондриальной Еве.
При этом нужно помнить, что генетические «Адам» и «Ева» -это своего рода фантомы, созданные масс-медиа. Под ними понимается скорее некая условная точка коалесценции всех существующих hic et nunc генетических линий; кроме этого, следует помнить они в определенном смысле асинхроничны и смещаются по временной шкале по мере вымирания той или иной генетической линии. Можно конечно вычислять общего предка до первейшего организма, только встает вопрос в самоценности такого анализа. А так — «генетические Адам и Ева» — весьма хороший символ общего происхождения человечества, особенно для современной евроцентричной цивилизации, во многом построенной на библейско-христианском символизме.
Теоретически можно стоить подобные филогении и по снипам в аутосомах. И хотя в последнее время много пишут про «атавистические» снипы и малорекомбинантные локусы в аутосомах, они все же не дают столь же убедительной филогении, как Y и mtDNA. Но мне кажется, что для людей, которые верят в происхождение современного человечества из Костенок, не имеют значения ни гомозиготность, ни гетерозиготность, также как и рекомбинантность/нерекомбинантность локусов.
Эксперимент номер 1
В течении периода между 2008-2010 гг., я усиленно занимался проблематикой филогенетики в применении к ДНК-генеалогии. Например, еще в далеком 2009 году я построил по известной выборке гаплотипов из статьи Роевера 2008 года «быстрое» дерево с гаплотипами гаплогруппами I. Модальные гаплотипы не вводил, корень получен мурковской (здесь и далее речь идет о замечательной программе Валерия Запорожченко MURKA) опцией MIDPOINTGROUP. Гаплотипы идут в том же порядке, что и гаплотипы I в файле у Рувера (название области +порядковый номер гаплотипа по области). Было найдено всего около 60 деревьев, из них я выбрал около 15 вариантов. Соотношение субкладов абсолютно иное, нежели в статье известного шарлатана Клесова, например I2b всего 8 штук (вместо 22 у Клесова), правда возраст клада такой же как у Клесова -в пределах статистической погрешности 10 000 лет. I2a всего около 70 гаплотипов.
Penzen-9
14,21,15,10,15,16,13,12,31,15,10,12,14,20,14,21,11
Smolen-2
15,23,15,10,15,15,14,12,32,15,10,11,14,20,15,21,11
Orlov-3
15,23,15,10,15,15,14,12,31,16,10,11,14,20,15,22,11
Nowgorod-3
14,23,15,10,13,17,14,12,32,18,10,11,14,20,13,21,11
Brian-4
15,23,15,10,15,15,14,12,30,16,10,11,14,19,16,22,11
Brian-5
15,23,15,10,15,16,14,12,32,17,10,11,14,20,22,21,11
Penzen-16
15,23,16,9,15,16,14,12,30,15,10,11,14,20,23,21,11
Tambov-6
15,24,15,9,15,16,14,12,30,16,10,11,14,20,14,21,11
Поскольку разрешение файлов огромное 9543*7672, то я не могу их выложить. Добавил всего 5 основных модалов из двух десятков, и результат оказался намного лучше, поскольку дерево было перестроено с учетом модальных гаплотипов всех субкладов I. Структура построенного дерева проиллюстрировала известный парадокс — хотя сама гаплогруппа I считается «старой», все ее густонаселенные ветви (или в терминах кладистики — клады ) — «молодые». Старых ветвей практически не осталось, что свидетельствует о древности I.
Возраст я не считал, хотя значение ро получилось равным 14 (а это достаточно много). И поскольку древо считалось быстро, значение 1 ро была взято равным 1000 годам. Возраст меня особо не интересовал, задача была другая — выявить в древе наличие трех ветвей. Кроме того, в ходе анализа я пытался определить место, где произошло разделение ветвей. Если самые старые ветвим I2 на Кавказе (допустим такой вариант), то разделение на I1 и I2 было именно там или раньше; но это не объясняет того, почему I1 в циркумкавказской зоне практически не видно. Видимо, c этих позиций популяционные генетики и рассматрыва Балканы, как предпочтительное место начала экспансии расселения I в Европу. Однако тут возможна масса альтернативных объяснений. Например, протопопуляция могла разделиться на 2 группы — первая ушла на Кавказ пошла, а вторая на Балканы. Или с Кавказа ушла часть людей, еще до разделения, и в ней произошло выделение линии I1.
Структура дерево выявила еще ряд актуальных проблемы, например гаплотипы I2b оказались гораздо ближе к корню по медианно сети, чем I1 и I2а. Этому имеется свое объяснение. В 2008 году уважаемый программист Вадим Урасин провел несколько недель в попытках доработать параметры median-joining таким образом, чтобы он корректно распределял гаплотипы в соответствии со снипами. Одним из способов улучшение топологии было использование так называемх весов маркеров. Однако, Поскольку расстояние между I2b*, I2b1 и I2b2больше, чем между I1 и I2a, все проги давали сбой.
В поисках решения проблемы я обратился к программе TNT, известной своей быстродействием.
Формат входного файла в TNT выглядит следующим образом
xread
‘optional title, starting and ending with quotes (ASCII 39)’
nchar ntax
Taxon0 1011110000
Taxon1 1111111000
Taxon2 1011110000
Taxon3 1111111000
;
Альтернативные «веса» маркеров, предложенные Урасином
DYS 393 0,00138 54
DYS 390 0,00235 50
DYS 19/394 0,00209 51
DYS 391 0,00164 53
DYS 385a 0,00252 49
DYS 385b 0,00372 46
DYS 426 0,00014 73
DYS 388 0,00103 56
DYS 439 0,00285 48
DYS 389-1 0,00194 51
DYS 392 0,00071 60
DYS 389-2 0,00397 45
DYS 458 0,00464 44
DYS 459a 0,00077 59
DYS 459b 0,00108 56
DYS 455 0,00028 67
DYS 454 0,00048 63
DYS 447 0,00345 47
DYS 437 0,00081 58
DYS 448 0,00150 53
DYS 449 0,00660 41
DYS 464a 0,00234 50
DYS 464b 0,00293 48
DYS 464c 0,00278 48
DYS 464d 0,00281 48
DYS 460 0,00251 49
GATA H4 0,00210 51
YCA IIa 0,00115 56
YCA IIb 0,00155 53
DYS 456 0,00376 46
DYS 607 0,00249 49
DYS 576 0,00593 42
DYS 570 0,00610 42
CDY a 0,00703 41
CDY b 0,00843 39
DYS 442 0,00299 48
DYS 438 0,00077 59
DYS 531 0,00063 61
DYS 578 0,00025 68
DYS 395S1a 0,00054 62
DYS 395S1b 0,00076 59
DYS 590 0,00017 71
DYS 537 0,00146 54
DYS 641 0,00051 62
DYS 472 0,000006 99
DYS 406S1 0,00198 51
DYS 511 0,00156 53
DYS 425 0,00132 54
DYS 413a 0,00287 48
DYS 413b 0,00244 49
DYS 557 0,00361 46
DYS 594 0,00067 60
DYS 436 0,00020 70
DYS 490 0,00032 66
DYS 534 0,00520 43
DYS 450 0,00029 67
DYS 444 0,00324 47
DYS 481 0,00478 44
DYS 520 0,00241 49
DYS 446 0,00361 46
DYS 617 0,00112 56
DYS 568 0,00087 58
DYS 487 0,00141 54
DYS 572 0,00132 54
DYS 640 0,00048 63
DYS 492 0,00052 62
DYS 565 0,00081 58
Программа TNT также принимает файл в формате Нексус. Для преобразования ych -> Nexus был написан конвертер уважаемого А.Лифанова. Изначально автором Мурки планировался свой собственный конвертер RDF <-> Nexus, однако по ряду причин он так и не был реализован. В этой связи стоит отметить, что TNT единственная программа наряду с MURKA, которая дозволяет вычисления по произвольной матричной метрике. В поисках обходных путей и в связи с трудностями с поддержкой форматов, мне пришлось воспользоваться командой prepare из дистрибутива MURKA (опции MX BINARIZE). С ее помощью, я сделал state matriх, немного отредактировал ее в родной формат .tnt, вставив файл снипы со значением 1 и 0, и загрузил ее в прогу. Затем возникал другая проблема, как сохранить в графическом формате филограммы, которые находит TNT. Я пробывал сохранять в .nex, tre и в метафайлы emf, однако они нигде не открываются. Я посмотрел формат .tre TNT, в отличие от читаемого Мегой файла с одним древом, в TNT-овском файле прописываются все древа.
Интересен тот факт, что формат в TNT, чем-то напоминает datamatrix в бинарном RDF, однако между ними нет совместимости. Вадим У. показал скрипт вызывающий Мурку, но, к сожалению, частая смена Валерием Запорожченко параметров сделала свое нехорошее дело. В скрипте потерялась опция UBRSPH — она не делает ничего нового, просто в последних трех версиях вызов с ней эквивалентен тому что было раньше без нее. Валерий также рекомендовал использовать BNDREPEATS2 и даже BNDREPEATS3 вместо BNDREPEATS1, но это уже дело вкуса и не столь жестко — эффект заметен не вcегда.
Эксперимент номер 2
После печального эксерзиса с построением «быстрого» дерева гаплогруппы I я несколько поостыл к методу MJ, и интуитивно склонился к методу Вадима Урасина. Тогда мне казалось, что если оптимизировать метод Урасина, то его применение откроет новые перспективы. Возможно даже, что реконструируемые с его помощью филогении кладограммы можно будет исторически объективно верифицировать за счет привлечения исторических и археологических данных. В ожидании усовершенствований программы, я воспользовался известным в биоинформатике программным пактом Арлекин (главным образом встроенным анализом AMOVA). Также, как и в прошлый раз, z не ставил в данном случае перед собой задачу выявления возраста I*, за значение одного ро в годах я брал некое приемлемо большое число (2000 лет), поэтому узел с возрастом в «45000 лет» в данном случае соответствует узлу последнего общего предка I* и J*. Меня больше интересовала задача, поставленная ув. А.Штруновым. Напомню,он попросил меня установить субкладную принадлежность 17 маркерных гаплотипов группы I с помощью филогенетических реконструкций. То есть, «разделить» ветви на дереве. Первая попытка была, как видно из вышенаписаного, неудачной. Поэтому я решил обкатать метод на более надежном 17 маркерном наборе модальных гаплотипов всей группы I. По способу, предложенному Урасином, ветви-клады «закреплял» введение в ych.файл SNP мутаций (через значений 1 и 0). Для большой надежности, использовал прием, который отлично себя показал на 67 маркерах, т.е укоренял через модал ближайшей к I гаплогруппы J.
Результаты подобного опыта лучше согласовались с данными номенклатуры субкладов I. Почти все субклады легли где им полагается (внутри родительских кладов), хоть и не в прогнозируемом номенклатурой порядке. Тем не менее, на 17 маркерах эксперимент не дал удовлетворительного результата даже после «читового» введения в ych. файл данных о снип-мутациях и применения «мощного» укоренения через предковый гаплотип соседней гаплогруппы J*. Из этого следуют следующие выводы:
1.База данных 17 микросателлитных YSTR гаплотипов, определенных по снипу, как I-M170, недостаточна для определения субкладовой принадлежности гаплотипов.
2. Без указания снипов и укоренения через ответвление другой гаплогруппы, выявленные топологии древа носят случайный характер.
3. Выделение ветвей I и I2a/I2b в 17-маркерной филогении даже после описанной выше «оптимизации» происходит способом, противоречащей данным о снип-мутационном схеме гаплогрупп I.
4. Принадлежность гаплотипов I в 17 маркерной записи к субкладу невозможно выявить с помощью методов филогенетических реконструкций. Это касается не только медианных сетей и МP деревьев Нетворка/Мурки, но и филограмм, построенных по методу NJ , Фич-Марголиаса и т.д.
5. Следовательно, «выделение молодой ветви и старой ветви» » гаплогруппы I2″ на 17 (sic!) маркерных гаплотипах, произведенное псевдоученым Клесовым в своей псевдонаучной статье о гаплотипах европейской части РФ, не является методологическим верным. В результате выявляются не реальные в филогенетическом смысле ветви, а некие фантомные объединения гаплотипов по некоторым мат.статистическим критериям. Таким образом, уже тогда Клесов показал свой дилетантизм на примере «разделения» I2a и I2b в своей статье о гаплотипах выборки Рувера. В свое оправдание, вместо четкого и грамотного ответа на вопрос, Клесов привел туманный и бессмысленный набор предложений общего характера: «мой подход — строить деревья гаплотипов, выделять и идентифицировать ветви, анализировать каждую ветвь отдельно, а затем сопоставлять базовые гаплотипы для всех ветвей и находить возрасть пра-предка. При этом получаются часто совершенно новые вещи. Техника счета — дело десятое, она уже отработана и выверена. Я просто выбираю нечетный индекс, как того требует программа. Я выбираю 9. Могу выбрать 7, 5, 3, другой — дерево перестроится, но останется по сути тем же.» Топология в Phylip нужна только для определения «выдающихся групп». О родстве между гаплотипами о них судит нельзя. Кстати, именно поэтому академик Клесов использует Филип и Мегу для «причесывания» выборки. Несмотря на настойчивые советы перейти на Мурку, ну или хотя бы для начала — Fluxus Network, так как в обеих программах используется концепция медианных сетей, — самозваный «академик» так и не смог перейти на следующий уровень. Мой спор с Клесовым начался из-за проблем с филогенетическим древом, реконструированном на основании 17-маркерных гаплотипов. Выяснилось, что «ветви» этого древа не соответствуют снип-разметке гаплогруппы I. В ответ на это Клесов посетовал, что я заранее загнал себя в тупик утверждением, что 17-маркерные гаплотипы ни при каких обстоятельствах не могут быть использованы. Действительно, в филогении — нет, не могут. Выше показывал примеры с цифрами и расчетами, почему это так. Для других целей — наверное можно хоть однолокусные брать. Смотря для чего. Например, в той же Меге можно найти 1000 разных топологий деревьев по одной и той же выборке. В MrBayes, с помощью байесовской интерференции — 50000 равновозможных (equiprobable ) филограмм, то есть деревьев 17-маркерной выборки. И каждая из них будет равноценной филогенетическим построениям Клесоа в Меге, поскольку, также как и у Клесова она не отражает объективной информации о сниповых партициях древа I.
В Mesquite есть хорошая опция сравнения частотности между ветвями консенсусного дерева и ветвями всего множества филогенетических политомных филогенетических деревьев. Так вот, средняя частота повторения отдельных ветвей во всех деревьях не превышает 0.0001-0.00005 (то есть, в среднем частота совпадения не превышает 0.01-0.005 процента от объема деревьева). Это свидетельствует о случайности разбиения ветвей, и ненадежности филогении. Все, что можно определить по таким деревьям — это визуально продемонстрировать удаленность/близость гаплотипов по матричной дистанции от корня. Кстати, совершенно неудивительно, что удаленность от корня значительна. Расчеты величины ро — среднего количества накопленных мутаций на каждый гаплотип — показывают, что в среднем на каждый гаплотип выборки по «мутационному» пути от «базового» гаплотипа накопилось 14-15 мутаций (из 17!). Поэтому и неудивительно, что гаплотипы сгруппировались подобным образом. Разбивать ветви и определять базовые гаплотипы по таким выборкам — то же самое, что строить медианные сети по раскладу карт Таро. Нам уже известно, что никакая программа не делает эту работу на 17 локусах хорошо. По прошествии 4 лет, Клесов так и не ответил на косвенный вопрос Валерия Запорожченко, заданный подчеркнуто мягко и ненавязчиво в ходе дискуссии: какова цель построения деревьев? Поиск предковых гаплотипов? Но при неправильном положении предков друг относительно друга это приведет к ошибке. Возраст всей I? Но зачем Вам для этого деревья? Может, просто кластеризация ветвей? Нельзя ли ясно указать, зачем запускалась программа? Фраза Клесова «программа рассматривает возможную последовательность перехода» имеет ясный смысл только если таких возможностей в принципе мало, ограниченное число. При гигантской гомоплазии 17 локусов вариант найденный Клесовым — один из миллиардов (и даже миллиардов миллиардов) потенциально возможных. Нет здесь «объективного отражения». На каком основании Клесов выбирает только один вариант, если возможна масса их, без дискуссии чем он лучше? Клесов сам говорил неоднократно, что применяемый им метод расчета возрастов зависим от разделения на ветви («считать чохом» vs «не считать чохом»).
6. Даже после построения дерева модалов I с использованием советов Урасина ( применение комбинированного метода, а такж вычисление стоимости байесовского древа ), cовершенно очевидна недостаточность 17 маркерных гаплотипов для построения филогении (в случае I гаплогрупп необходимо минимум 30-35 маркеров), о чем собственно писали Валерий З. и Вадим У.
Здесь нужно сделать важное замечание. Достаточность или недостаточность длины гаплотипов диктуется исключительно целями исследования. Ведь любой вменяемый человек понимает, что надежное и достоверное парсимонистское или байесовское разделение гаплотипов (без знания субклады и соответствующего снипа) в соответствии с истинным мутационным древом затруднительно и на выборке гаплотипов с бОльшей, нежели 67 маркеров, длиной гаплотипной записи. Однако если выборка сформирована по генеалогическому способу (как например в исследованиях по ирландским кланам) или в выборках однофамильцев (те же исследования ирландцев о корреляции фамилии/гаплогрупп, проект «Однофамильцы» и так далее), то 17 маркерных гаплотипов достотаточно для кластеризации и оценки статистического соответствия фамилий и гаплогруппы. Однако для генеалогического масштабирования/позиционирования ветвей в филогенетическом дереве и большего количества маркера недостаточно, так как выявляемые ветви носят случайный характер, особенно когда выборка «чоховая», то есть включает несколько кладов или субкладов гаплотипов общей гаплогруппы. Все что можно сделать — это кластеризовать в филогенетечиском древе/сети наиболее устойчивые гаплотпы (правда, это можно сделать и без построения сетей) и при этом помнить, что из-за мощной гомоплазии таких выборок (например в той же руверовской выборке I, где среднее количество накопленных мутаций от предкового гаплотипа составляет 14 мутаций из изначальных 17), в кластеры неизбежно попадут «посторонние» гаплотипы. Затем нужно выделять те ветви, которые особенно выделяются и по ним заново строить дерево, и в больших выборках нужно делать так так до самого мелкого уровня. В этом случае надо увеличивать не значение эпсилона (он нужен для оценки гомоплазии, то есть сколько параллелизмов в сети), а использовать опцию Post-processing>MP. Тогда можно в структуре сети выявить деревья — которые грубо говоря, представляют собой кратчайшее расстояние между узлами сети (то есть междк реальными гаплотипами, которые в исходном файле обозначены фамилиями или номерами, а также «реконструированными» гаплотипами «предков», они обозначены как mv). Есть еще один метод, обратный вышеописанному. Он называется bootstrap analysis, только в нем не добавляют гаплотипы в выборку, а наоборот, изымают из выборки произвольные гаплотипы. Этот метод реализован, в частности, в одной из филоутилит в пакете Phylip. С его помощью оценивают надежность той или иной ветви древа. В 2009-2010 годах я каждый день занимался подобным методом на практике с реальными гаплотипами.
Примечание. Для понимание некоторых технических деталей необходимо вкратце объяснить суть методов программ. Например, в Phylip при построении деревьев используется один из трех дистантно-матричных методов: метод Фитча-Марголиаша, метод ближайшего соседа или метод с использованием группировки. Как правило, применяется метод Ф-М или метод «взвешенных» минимальных корней. В отличие от метода ближайщего соседа, где в качестве метрики используются та самая матрица генетических расстояний (которую любители ДНК-генеалогии получают в YUtility) Этот метод удобен для кластеризации «близких» гаплотипов, так как в расчет принимаются линеарные генетические расстояния (дистанции), таким образом, что наиболее близким гаплотипам присваиваются более высокие «веса», тем самым понижается степень неаккуратности расчета измерения дистанций между более удаленными гаплотипами. Хотя, как это не странно, бывает и так, что не-взвешенное дерево ближе к верифицируемому с точки зрения генеалогии древу. Расчет ведется от первого гаплотипа, которым в YUtility является условный предковый гаплотип — так называемый модальный гаплотип. Предполагается, что этот модальный гаплотип (или гаплотип с медианными значениями соответсвующих маркеров всех гаплотипов выборки) совпадает с гаплотипом «общего предка», хотя на практике это зачастую не так. Я как то специально проверял эти модальные гаплотипы в Мурке на предмет их совпадения с филогенетическим «мидпойнтом», и в половине случаев гаплотип мидпойнта и модальный гаплотип не совпадали. Часто встречающийся у новичков конфуз с ветвьями дерева вызван тем, что новички путают понятия генетической дистанции и сходства гаплотипов. Дистанция считается в этом случае от модала, например гаплотип A ближе к модальному гаплотипу (то есть в нем накоплено меньше мутаций от предкового гаплотипа, и поэтому дистанция меньше), гаплотип B — мутаций больше и поэтому он дальше. Это вовсе не исключает вероятность того, что по числу совпадающих маркеров B ближе к A и, например, к C. В этой связи необходимо напомнить определение гомоплазии: гомоплазия — однотипная изменчивость признаков и свойств у организмов различных таксономических групп при параллельных, но независимых эволюционных процессах. То есть тут идет речь о возможных параллелизмах, когда (в применении к Y-STR анализу на мельчайшем, близком к генеалогическому уровню филоанализу) в параллельных ветвях могут возникать сходные признаки ( в нашем случае — одинаковое число аллелей в локусных маркерах), при этом их генеалогическое родство по патрилинейной или если угодно -игрек-хромосомной линии — намного глубже.
Эксперимент номер 3
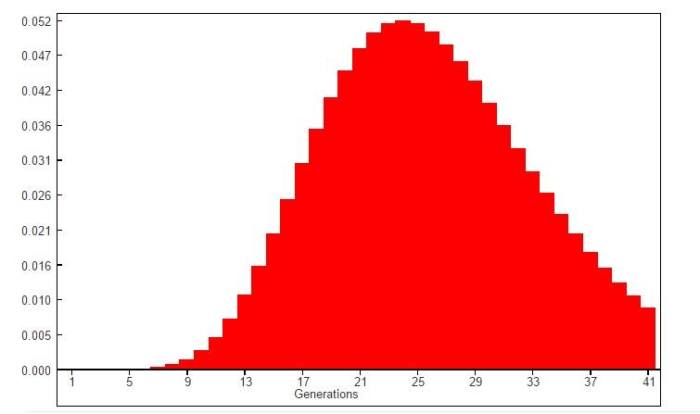
В том же 2009 году я открыл для себя новую замечательную программу — MrBayes -программу для анализа филогений с помощью байесовской инференции. Так как программа понимает формат Nexus, то я конвертнул поправленный ych файл в формат nex, немножко поправил файл согласно примерам, и запустил марковскую цепочку по методу Монте-Карло. Рабочий цикл программы прошел нормально, но медленно — на 145 гаплотипов примерно 3500 циклов за 20 минут. На выходе после «прожига» деревьев были получены деревья в формате Newick. На графике соотношения RI (индексов ретенции) между выявленными в MrBayes 2100 (!) вероятными деревьями группы из 145 43-маркерных гаплотипов, cреднеарифметическое значение индекса составило: 0.17967889; медианное: 0.17454545 (n=2001 деревьев). Под индексом ретенции в кладистике понимается доля очевидной синаморфии значений (в нашем случае локусных значений), которая присутствует в качестве синаморфии в построенном дереве). Формула расчета: RI= (g-s)/(g-M), где g-верхний порог значения, принимаемого локусом в любом построенном дереве, s- минимальное количество шагов изменения значения локуса в дереве, M-число шагов которое требуется в конкретном дереве.
R моюму удивлению, полученные деревья относительно быстро загрузилась как в SplitsTree, так и в Меге и TreeView (причем, в Меге они грузились дольше всего). Есть такая фича во всех этих программах (а также Mesquite), которая позволяет строить по всем имеющимся вариантам «компромиссное» consensus-tree по двум разными методикам (в том числе и UPMGA).
MrBayes еще хорош тем, что высчитывает апостериорные вероятности каждой ветви каждого древа. Для 17 маркерной выборки вероятность «самого точного» дерева (то есть при SD < 0.001) не превышает 53%.
К концу 2009 года я начал склоняться в пользу более сложного трехступенчатого анализа:
1)Постройка всех возможных вариантов деревьев и оценка их вероятности с помощью байесовской инференции (программа Mr.Bayes). Затем сортировка наиболее вероятных деревьев и их визуализация в Меге, СплитсТри, ТриВью, Мескит. Оценка каждого наиболее вероятного дерева, а также консенсусного дерева этих вариантов. Предварительная группировка и предпредварительная корректировка с использованием знаний о генеалогических группах, а также с данными о вероятности той или иной ветви. Дальше нужно смотреть мутировавшие позиции, и если подходить к вопросу методично, просчитывать, какое из двух деревьев более вероятно. Тут лучше всего использовать байесовский метод. То есть брать абсолютные скорости мутаций и перемножать их на скорости (точнее вероятности) не-мутаций.
2) MJ-анализ каждой группы отдельно, постройка сети с использованием маркерных «весов», затем нахождение субоптимальных и MP деревьев в медианной сети (Murka). Анализ полученных результатов с использованием гипотез о генеалогических группах, использование данных о партициях дерева и т.д.(статистические данные по деревьям в MURKA)
3) Расчет с помощью Байеса по данным о априорных вероятностях n-мутациях (n*число маркеров*вероятность мутации локуса*вероятность немутаций) и по данным о взвешенных партициях (апостериорная вероятность ветви). Выбирается наиболее вероятный результат. Данные сверяются с генеалогическими (историческими, генеографическими и т.д.) данными.














{kind=link}
Для отправки комментария необходимо войти на сайт.