Выполняю с небольшим опозданием данное ранее обещание и расскажу о слабых местах выявления процентов этнического происхождения с помощью анализа результатов ДНК-тестирования. Последние лет пять этот тип изучения этно-популяционного происхождения с привлечением данных генетики вошел в моду — в 2011 году, когда я первый раз провел подобный анализ существовало все 2 крупные компании в пакет клиентских услуг которых входило проведение подобных анализов клиентских данных. Ровно столько же было заметных в инете любителей, предлагающих более развернутый и разжеванный вариант подобного разбора этнопроисхождения добровольных участников своих проектов. Главным инструментым и тех и других являлись программы типа Admixture и STRUCTURE (разработанные академическими биоинформатиками для решения одной из задач популяционный генетики — а именно определения этнической субструктуры в структуре изучемых в ходе конкретного исследования национальной или региональной выборки народонаселения).
Прошло лет 6, я провел более тысячи подобных экспериментальных анализов — на принципиально разных выборках и образцах аутосомных снипов представителей разных народов. Каждый из таких экспериментов хотя бы немного отличался от других — и не только числом заранее заданных предковых компонентов этнических популяций, но и разнообразием самих этих популяций, их числом и качеством генетипированных в этих популяциях снипов, — например степенью сцепления снипов между собой, процентом минорных вариантов, количеством снипов, соотношение гомо- и гетерозиготных аллельных вариантов. На первом этапе основной проблемой являлась недостаточная представленность отдельных этносов в выборке вкупе с неполным совпадением популяций по числу генотипированных снипов
Позднее я частично научился обходить проблему за счет импутирования аллельных вариантов недостающих (негенотипированных) снипов по большим референсным панелям. В частности используемый Сергеем Козловым калькулятор K27 был сделан мною как раз с использованием таких импутированых вариантов.
Еще позже — после прорыва в области изучения палеоДНК — появилась возможность не угадывать предковые компоненты слепым перибором числка K (предковых компонентов), а задавать заведомо предковые популяции людей (жителей мезолита, неолита и бронзового века) в качестве чистых предковых популяций К современного народонаселения. Таков, например мой этнокалькулятор K11 Ancient, загруженный зимой этого года на Gedmatch.
Всего же за это время я разработал не менее 60 разных моделей в интервале от K=7 до K33, причем для многих K я разработал сразу несколько моделей.
Все эти модели (только калькуляторы; без инструментов поиска ближайших к тестируемому популяций) я размещаю в открытый доступ на OneDrive (ссылка открывается при нажатии на картинку). Эти файлы работают с программой DIYDodecad, инструкцию использования которой можно найти на сайте Диеникиса
Я решил подвести итог этому направлению своей деятельности, на которое ушло много сил, cредств и почти все мое свободное время. Вместо этого я переключусь на более точные формальные методы определения этнического происхождения, разработанный в генетической лаборатории Дэвида Райха из Гарварда.
Главная причина — в силу своего перфекционизма я не был доволен точностью определения частот конкретных предковых компонентов в состав генома отдельно взятых людей. Кроме того, этой зимой в ходе бесед с подобным же любителем насчет проблем Admixture, мы обнаружили ряд причин, приводящих при анализе данных отдельно взятых людей к странным и заведомо неверным комбинациям этнического раскалада предков.
Есть еще одна причина — перенасыщение данного маленького сегмента на рынке инетрпретации генетических данных. В настоящий момент существует уже целый ряд компаний (не менее дюжины), вышедших на рынок ДНК-генеалогии в относительно недавнее время. Каждая из них разработала свой алгоритм и красивый графический интерфейс для визуализации данных по прогнозируемому этнопроисхождению клиента. Увеличилось число крепких и активных любителей (я знаю не менее 10 таких людей), занимающихся в принципе тем же самым, причем иногда качества полученных ими моделей этнического происхождения выше таковых в коммерческих компаниях. Благодаря их усилиям, число доступных этнопопуляционных калькуляторов увеличилось буквально в разы.
Но перейдем к конкретике. Часто люди систематически получают странные результаты — таких примеров можно приводить много. Причем иногда такие странные и неверные расклады можно встретить в больших этнических сообществах — например у чеченцев стабильно в MyHeritage выскакивают в раскаладе предковых групп одинаковые 10-15% жителей Британских остров.
Этот, конечно, нелепый пример, отлично иллюстрирует первую проблемы, связанную с разделением выборки и клиентской базы на кластеры. В отличии от любителей; большинство коммерческих компаний (за исключенеим разве что FTDNA, где алгоритм опеределения процентов этнического происхождения разработал как раз любитель) не занимаются поисками настоящих предковых компонентов — вроде европейских охотников-собирателей, земледельцев и скотоводов бронзового века. Вместо этого все образцы популяций — преимущественно из академических источников — объединяются по географическому признаку в отдельные кластеры — например скандинавский или балканские кластеры. Кластеры задаются как условные предковые компоненты (их может быть довольно много — как например в компании AncestryDNA), якобы позволяющие в данном случае более точно выявить недавнее этнопопуляционное происхождение клиента. И что хуже всего в эти же кластеры включают данные самих клиентов — очень часто просто со слов клиента о своем этническом происхождении (как было в своем время в 23andme), хотя в последнее время в некоторых компаниях (AncestryDNAO) алгоритм усовершенствовали путем включения дополнительных фильтров для отсеивания (например с помощью определения в анализе главных компонентов резко отличающихся и резко выделяющихся в плане этнического происхождения клиентов). Тем не менее, даже самое методичное применение всевозможных дополнительных фильтров не может гарантировать повышение точности предика этнического происхождения. Проблема что в человеческих популяциях — за исключением небольшого количества изолированных задрейфованных популяций вроде нганасан, чукчей, ульчей, калашей, папуасов — ни в одной из этнических групп компоненты не являются дискретными, а представляют собой градиенты частот аллелей, очень часто с большим расбросом из-за чего хвосты частот распределния этих частот перекрываются. На практике этот феномен приводит к появлению в индивидуальных клиентских данных фантомных компонентов — например у европейцев часто появляются всевозможные невозможные компонентоы происхождения — Amerindian, Papuan, Onge и так далее. Подобный подход только вносит сумятицу или — говоря статистическим языком — шум в результаты.
Очевидно, что данная проблема связана с классической проблемой статистики — проблемой организации и подразделения выборки. Схожей по характеру проблемой являеется проблема разнообразия выборки используемой для определения компонентов происхождения. Очевидно, что очень сложно впихнуть все генетическое разнообразие человечества в относительно небольшую выборку — даже еслии ее размер достигает полмиллиона или больше образцов (как у 23andme). Проблема в сверхпредставленности отдельных этнических или квазиэтнических групп в подобных базах данных (западных европейцев, американцев, финнов, ашкеназов и так далее). При неравномерности выборки наблюдается другой классический статистический эффект — искажение результатов выборки в сторону наиболее представленных групп (как было в свое время в 23andme, когда наблюдался эффект сверхпредставленности евреев-ашкеназов в количестве так называех генетических совпаденцев).
Еще одна схожая проблема — в количестве совпадающих снипов (одинакового числа снипов) между тестируемыми индивидуальными образцами и референсными группами. Это проблема затрагивает, правда, только калькуляторы разработанные любителями на базе DIYDodecad — в алгоритмах коммерческих компаний число снипов в рефренсных популяциях и индивидуальных образцах одинаково, т.к. анализируются только те образцы, которые тестировались самой компанией. В вышеупомянутых же этнопопуляционных калькуляторах анализируемые всегда «кроссплатформены» — и если разработчик использовал для разработки калькулятора только те снипы, которые содержаться в чипах 23andme, тогда при анализе данных полученных в компаниях FTDNA или AncestryDNA совпадением снипов будет частичным (так как снипы генотипируемые в этих компаниях совпадают лишь частично). В итоге ситуация в которой сравниваются аллельные частоты снипов референсных популяций (полученные при одном количестве снипов) с аллельными частотами снипов индивида (полученные при совсем другом количестве снипов). Элементарная логика подсказывает что в таком случае будет наблюдаться искажение результатов в совершенно непредсказуемую сторону.
К счастью, у обеих проблем есть разумные решения. Число совпадающих снипов между чипами FTDNA, 23andme (разных версий) и AncestryDNA составляет примерно 300 000, что достатчно для создания калькуляторов приемлемых для анализа данных от всех этих компаний. Решение первой же проблемы тоже есть, но его стоимость немыслимо выскоа — необходимл использовать примерно несколько десятков миллионов ПОЛНЫХ геномов популяций людей со всего мира. Разумеется, никакие любители никогда в жизни не смогут собрать такое количество данных необходимых для создания сверхточных калькуляторов.
Все вышеперечисленные проблемы — сущая мелочь в сравнении с настоящими проблемами, обусловленными алгоритмической стороной вопроса. Дело в том, что все компании (и разумееися любители) — так или иначе — при вычислении аллельных частот в компонентах референсных популяций используют программы использующие парадигму Admixture/Structure. А они используют ML-алгоритмы, минимизирующие ГРУППОВЫЕ частоты аллелей между всеми образцами выборки, т.е. аллельные частоты ПОЛНОСТЬЮ зависят от состава исходной выборки, даже в случае так называемого supervised («обучаемого») анализа, в ходе которого некоторые популяции принимаются за исходные «чистые предковые группы». Поскольку в ранних версиях Admixture, отсутствовала опция фиксирования одной из вычисляемых матрицы (P- матрица аллельных частот снипов в каждом из гипотетических компонентов происхождения; Q-матрица — матрица индивидуальных коэффициентов вклада предковых компонентов в индивидуальный геном с общей построковой суммой в 100%), и практически все компании использовали один и тот же алгоритм (он в во всех подобных программх схож — хотя разняться его имплементации и способы оптимизации функции правдоподобия), то все они подвергнуты искажению истинных частот аллелей. Этот косяк вносит решающий вклад в появление фантомных компонентов происхождения.
То, что вычисленные таким способом значения (скажем русского) могут очень сильно отличаться в сравнении с индивидуальными частотами аллелей (для примера такого же русского из той же скажем Орловской области) — было впервые замечено геномным блоггером Polako.
К сожалению, в силу своем личной ненависти к первоначальному разработчику DIYDodecad — греку Диенекису Понтикосу — он не смог дать формальное объяснение феномена и назвал этот феномен «эффектом калькулятора» (как бы намекая на косорукость кода этой утилиты). На самом деле сам калькулятор здесь не причем — все дела в приниципиальных различиях между определение происхождения на основании частот аллелей вычисленных по группе образцов и тем же самым вычислением аллелей, но уже не в группе, а в индивидуальныом порядке. В этом легко убедиться самому — возьмите клиентские данные, например, норвежца. Вставьте его в большую выборку образцов (например 2000 человек) и прогоните в программе ADMIXTURE задав такое количество гипотетических предковых популяций (K), при котором становится заметна субструктура генофонда популяций на внутриконтинентальном уровне. А затем возьмите того же самого норвежца, но уже в единственном числе, и зафиксировав полученные в предыдущем шаге аллельные частоты в виде предковых популяций. Вы увидите, что различия между результатами анализа одних и тех же данных могут достигать 20 а то и более процентов. Это-то и есть ядро так называемого пресловутого эффекта калькулятора. Очевидно, что и Оракул (т.е. инструмент определения ближайших к клиенту этнических популяций а также моделирования происхождения клиента через набор из 2, 3, 4 популяций) в этом случае (искаженных аллельных частот) будет искусственно создавать фантомные предковые популяций. Например, у русского из Владимирской области могут появиться в качестве шведы,
эстонцы или англичане из Кента.
Строго говоря, первым написал об этой проблема некий Vikas Bansal — автор программы iAdmix:
«For comparison, we also ran ADMIXTURE (in supervised mode using the HapMap reference panel of individuals) on the same dataset (see Figure 1(b)). The European and African admixture estimates for each individual were highly consistent between the two methods. For some individuals, the European component of ancestry using our method was split between the TSI and CEU populations. This could reflect one important difference between the two methods in how they use data from reference individuals. Our method finds a maximum likelihood estimate of the admixture coefficients for each individual using the fixed set of allele frequencies. In contrast, ADMIXTURE, in the supervised mode, utilizes data for all individuals (both the reference populations and the individual(s) being analyzed) to estimate the allele frequencies for each cluster or population and maximize the likelihood function summed across all individuals. Therefore, the allele frequencies are determined not only by the genotypes of the reference individuals but also by the individual(s) that are analyzed for admixture. To confirm this, we estimated allele frequencies by running ADMIXTURE twice: (1) using 800 reference individuals simulated using allele frequencies for 8 HapMap populations (100 individuals per population, see previous section) and (2) 800 reference individuals and 1 additional individual with 100% CEU ancestry simulated using the HapMap allele frequencies. Subsequently, we used our method to estimate admixture coefficients for the simulated CEU individual using the two sets of allele frequencies separately. We found that using the first set of allele frequencies, the admixture coefficients for both CEU and TSI were non-zero. In contrast, using the second set of allele frequencies, only the CEU admixture coefficient was non-zero. This was similar to the results observed in the analysis of the Mozabite data and provided an empirical validation of our hypothesis regarding the difference in the admixture coefficients estimated by the two methods.»

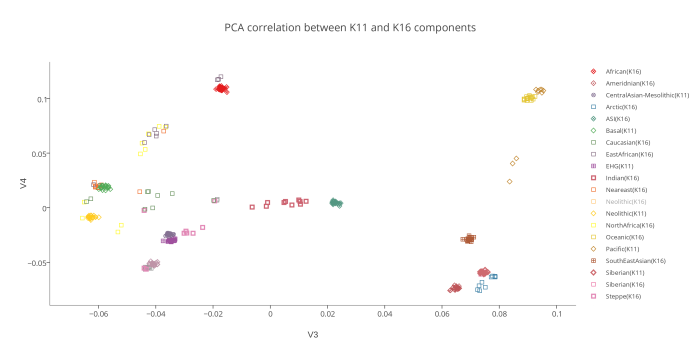 Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).
Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).
















































 )
)














Для отправки комментария необходимо войти на сайт.