**
Разработчики pyGenClean разместили полезный инструмент для предварительной подготовки выборки популяций для GWAS и этно-популяционного анализа. С помощью можно значительно автоматизировать относительно сложный процесс нахождения генетических outliers (т.е посторонних образцов выделающихся на фоне гомогенной однородной структуры популяции), а также провести многомерное шкалирования имеющихся популяций.
**
Я закончил проект по изучению структуры аутосомного генофонда грузинских этнографических групп. Ниже приведены выполненные в проекте публикую графики c результатами многомерного скалирования (MDS) и анализа главных компонент (PCA) в изученной выборке. Еще я понял свою главную ошибку во время работы с предыдущими графиками — она состоит в том, что я раньше не сохранял в R framework данные и историю проделанных над ними операций. R очень гибкая среда для статистического анализа, но в силу большого разнообразия существующих пакетов для визуализации данных для выполнения одних и тех же команд часто возникает путаница с выбором подходящей техники визуализации. Поэтому лучше всего не начинать каждый раз с нуля, а сохранять workflow для последующих экспериментов. 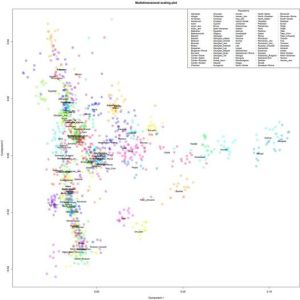

**
В русскоязычном секторе Интернета увеличивается число простых людей (и не совсем простых людей, вроде Татьяны Толстой), которые не боятся рассказывать открыто о своих генетических рисках, хотя в силу своего непонимания того что именно означает указанная в отчете risk odd (вероятность риска) , многие их выводы выглядят наивными.
Впрочем, ничего нет нового под Луной. Многие из моих сверхоптимистеских собеседников предполагали, что именно благодаря 23andme у рядового обывателя появилась возможность наблюдения за своими генотипами (или геномами , под которым мы — summa summarum — понимаем здесь всю совокупность прочитанных генотипов), и даже за динамикой экспрессии свого экзома.
Тем не менее, даже я помню, как задолго до начала моего увлечения генетикой, примерно в 2002 году я видел передачу про исландскую компанию Decodeme по Discovery Channel. После длинного интервью с тогдашним ведущим сотрудником этой компании (К.Стефансон), в котором он рассказал о тотальном (почти 80%) генотипировании всей исландской нации, создатели фильма взяли краткие интервью у простых исландцев. Мне запомнился один исландец-докер, который — не отрываясь от процесса разгрузки траулера с рыбой, — с улыбкой на лице сказал: «Я могу выпивать по 10 чашек кофе в течении одного часа. Cогласно исследованиям ученных из DeCODE Genetics, в гене метаболизма кофеина у меня аллельный вариант, повышаюший скорость метаболизма кофеина».
Вывод — 23andme не были первыми, их заслуга в другом — в том что они вывели персональную геномику (в ее упрощенной форме) на новый, международно доступный уровень.
**
Компания Nanoporetech выпустила на рынок портативное устройство MinION, предназначенное для анализа молекул (в том числе и молекул ДНК), его можно применять для анализа структуры протеина и секвенрования ДНК. Устройство можно подключить к обычному компьютеру через USB-порт.
**
Уважаемый Pavel Bernshtam предложил реалистичную перспективу на стартапы. Кроме всего прочего, между строк замечаний Бернштама можно прочитать имплицитное неявное объяснение феномена значительной молодости самых известных стартаперов (им нечего терять и их руки-головы не связаны-загружены семейными обязанностями прокормки супруги и спиногрызов).
Я стою на перепутье выбора между развитием идеи этно-популяционного ДНК-калькулятора в форме стартапа, либо форме краудсорзинга, либо некоммерческая инструментализация разработки в криминалистике (в виде патента на методику нового вида криминалистической ДНК-экспертизы, которая со временем заменит надоевший всем фбр-овский CODIS):
«Хорошо, если просили про стартапы. Для стартапа нужно несколько вещей. Самое простое — идея. Идея сама по себе не стоит ничего. 0. Самая классная идея — НИЧЕГО. Идея начинает хоть что то стоить (тоже немного) если на ее основе написан бизнес план. Обоснованный бизнес план. Бизнес план, который может убедить. Сколько юзеров придет к вам на сайт в первые полгода? миллион? А почему? Докажите. А сколько зарегестрируется? Почему?
Следущее, что нужно — человек, который может принести инвестиции. Для этого нужно — представительность, бизнес план, знакомства и уйма всего иного. Нужно найти выход на инвесторов (без выхода тоже можно, но разговаривать с тобой будут иначе), нужно что бы тебя порекомендовали, нужно уметь рассказывать и убеждать. Далее — деньги. Скорее всего у Вас не получится сделать прототип, достаточный для получения инвестиции вечером на коленке, параллельно с основной работой. Вам надо будет уволиться и писать код.»
**
Как Вы помните, на Gedmatch.com были размещены разработанные мною этно-популяционные калькуляторы MDLP на платформе DIY Dodecad. Они позволяют довольно-точно определять этническое и популяционное происхождение исходя только из сравнительноого анализа частот полиморфизмов ДНК протестированного человека с частотами полиморфизмов ДНК в референсных популяциях. Несмотря на простоту использования (загрузил свое raw data, нажал на кнопку — получил результат), основные пользователи этого инструмента — американцы — имеют траблз с пониманием и интерпретацией результатов. Вот например, из свежего, присланного мне в январе. Ко мне уже обращаются как к доктору, который должен выдать свой авторитетный этнодиагноз:
» I had my test at 23and me and it has me as 100 European.
My mom says its a lie as my dad was an inuit from Alaska .My kit is ******
Could you please debunk inuit story»
Papa was a rolling stone (c)
«My results are for North-Amerind, (North American Indian) .. I suspect 4 generations back
Chr 1 1.7%
Chr 7 3.3%
Chr 18 2.5%
Is this a definite result for American Indian Heritage?»
На такие письма я вообще больше не отвечаю. Весьма странно что у столь многих американцев в последнее время появился фетиш происхождения от американских индейцев. Раньше это было не так заметно.
**
Повторное ресеквенирование «древнего» генома останков жителя мезолитической Иберии из La Brana 1 (того самого, которого исследовали в позапрошлом году на аутосомы и митохондриальный геном) показало, что этот человек имел очень необычную для Европы Y-хромосомную гаплогруппы — С6. Странности заметны на и уровне фенотипа: согласно анализу комплекса снипов, определяющих на уровне генотипа цвет кожи и глаз, он был темнокожим человеком с голубыми глазами (!). У древнего европейца, жившего в пещере Ла-Бранья-Аринтеро (La Braña-Arintero, León) на севере Испании примерно 7 тысяч лет назад, были голубые глаза и очень смуглая кожа. Так художник представил себе то, как выглядел житель испанской пещеры 7 тысяч лет назад. (Ниже рисунок, опубликованный в Эль Паис.)

Палеогенетики успешно прочитали ДНК из костей древнего европейца, жившего в одной из пещер на севере Испании примерно 7 тысяч лет назад, и выяснили, что у него были голубые глаза и очень смуглая кожа, говорится в статье, опубликованной в журнале Nature. «Главным сюрпризом для нас стало то, что этот человек обладал типично «африканскими» версиями генов, которые управляют пигментацией кожи, что вероятно делало его очень смуглым или даже темнокожим, хотя мы и не можем точно определить ее тон. Еще более удивительным стало то, что этот «испанец» обладал теми вариациями генов, которые делают глаза европейцев голубыми, что делает этот геном уникальных, так как по всем остальным признакам он происходит из Северной Европы», — заявил Карлес Лалуэса-Фокс из Института эволюционной биологии в Барселоне (Испания). Что касается редкой гаплогруппы (C6, или по мнению некоторых исследователей просто C), то оказывается, что еще в 2013 году несколько любителей-непрофессионалов предсказывали вероятность присутствия С у части жителей палеолитической и мезолитиской Европы — по их мнению, мужское население палеолитической Европы могло принадлежать к линиям — C-V20 (в ISOGG С6), F и IJ.
«Ранние представители современного человека в Европе (EEMH), широко известные как кроманьонцы, мигрировали с Ближнего Востока в Европу несколькими волнами. Задумывашись над тем, какие гаплогруппы Y-ДНК могут быть связаны с ними, и в каком порядке они мигрировали в Европу, я придумал следующую хронологии для верхнего палеолита.
1) Гаплогруппа С6 (или С *, которая развилась в C6 в Европе)
2) Гаплогруппа F
3) Гаплогруппа IJ (которая развилась в Европе в гаплогруппу I) «
Заслуживает внимание и мастерское использование в данном исследовании методов секвенирования нового поколения — в частности, после того как генетики собрали геном древнего европейца из прочитанных мелких сегментов ДНК («ридов») по методу отображения ридов на референсный геном человека, осталось приличное количество неиспользованных ридов. Генетики использовали «сухой остаток» для проведения метагеномического анализа. Как известно, метагеномика работает с набором всех ДНК находящихся в среде; следовательно генетики сделали удачное предположение о том, что «риды» без привязки к человеческому геному принадлежали геномам бактерии. BLAST-анализ ридов в Генбанке позволил установить те виды бактерий, секвенсы геномов которых были наиболее близки к изучаемым ридам.
В конце января были опубликованы две замечательные статьи на русском языке, посвященные бурно развивающейся области исследований — молекулярной патологии: «Молекулярная патология и роль врача-патологоанатома» и «Наследственно обусловленный рак молочной железы и яичников«.
The Coop Lab продолжает размещать материалы о статистических рассхождениях в характере наследования генетического материала у ближайших родственников. Традиционно считается, что сибсы (сиблинги) одного пола похожи друг на друга в той или иной степени. Различие в фенотипических чертах объясняются разными факторами окружающей среды воздействующих в разной степени на их развитие. Тем не менее, как было показано в статье The Coop Lab,сибсы различаются также на уровне своего генома, за счет случайности сегрегации и рекомбинации.
Китайские генетики разработали новый метод генной хирургии (точное геномое редактирование) и успешно применили его на макаках.
Ученные из университета Северной Аризоны «возродили» вирус древней чумы, пандемия которой пришлась на время правения византийского императора Юстиниана (Юстинианова чума). В лаборатории был прочтена последовательность ДНК бактерии-возбудителя чумы, которая содержалась в останках жертв этой пандемии. Очевидно, здесь также применялись методы метагеномики.
В сетевой версии журнала «Наука и жизнь» размещена статья о характере генетической интрогрессии (межвидовым обменом чужеродной генетической изменчивостью) произошедшей между неандертальцами и предками анатомически современного человека много десятков тысяч лет назад, и приведшей к частичной гибридизации двух видов, чьи эволюционные пути разошлись около полумиллиона лет тому назад:
«Оказалось, что практически все неандертальские гены локализованы в Х хромосоме, а значит, передались нам по женской линии. Ученые пришли к выводу, что мальчики, рождавшиеся в результате смешения кровей, были в большинстве своем бесплодны. «Когда неандертальцы и люди скрещивались, это было на краю биологической совместимости, ведь два генома не встречались друг с другом примерно полмиллиона лет», — комментирует результаты исследования один из его авторов Дэвид Рейч, генетик из Медицинской школы Гарварда (США).»
Я еще в 2010 году говорил, что если смешивание с неандертальцами происходило, то скорее всего гены были привнесены от связей между мужчинами homo sapiens sapiens и женщинами-неандертальцами. Не откажу себе в удовольствии процитировать свое сообщение на форуме Молгена.
«Re: Люди носят гены неандертальцев
Ответ #23 : 10 Май 2010, 19:40:25 Самое неубедительное в обеих работах это
1)отбор снипов для анализа (перекрестное сравнение снипов орангутанга, человека и шимпанзе — выбрали те, которые у человека являются, как считается, потомковыми).
2) по отобранным снипами произвели выравнивание (alignment) секвенсов шимпанзе, человека и неандертальца фазирование предкового генотипа общего предка человека, неандертальца и современного человека (т.е говоря проще, реконструировали (предсказали) гипотетический генотип по методу Байесовской апостериорной вероятности)
3) затем разбили фрагменты генома неандертала по снипами по признаку совпадения или несовпадения с предковыми значения гипотетического секвенса общего предка шимпанзе и гомо, на три группы -гомозиготные с предковым значением снипа, гомозиготные с потомковым значением и просто гетерозиготы. Про исключение более половины мутаций (пусть и синонимических), я вообще молчу. Но кто может гарантировать, что предковый генотип реконструирован верно, и, что самое главное — где доказательство того, что у неандертала должно быть именно предковое значение снипа, а не мутировавшее параллельно с человеком.
Наконец, на приведенном выше графике, разброс участков генома совпадающих у человека и неандертальца по X хромосоме, находится в меньшем диапозоне SD (стандартного отклонения), эти участки небольшие, но по структуре более дивергентные.
Из чего следует 2 вывода:
a) основное генное вливание шло через X хромосому и b) поскольку около 2/3 генетической информации X хромосомы аккумулируется в женских линиях, то направление вливания шло через самок неандертальцев и мужчин-сапиенсов, что несколько противроечит картине изображенной в первой статье.»
Любопытно, что при ресеквенировании геномов неандертальцев и секвенировании геномов новых неандертальцев (из пещеры Окладникова) применили новый метод секвенирования. В частности, они секвенировали митохондриальную ДНК из кости неандертальца и отделили ее от ДНК современного человека, что позволило доказать родство между жившими в Сибири и в Европе неандертальцами.Метод определения посторонних наслоений ДНК основан на анализе ее естественных мутаций. Так, у 30–40% образцов, возраст которых насчитывает несколько тысяч лет, цитозин превращается в тимин, а гуанин — в аденин. Ученые разработали систему, моделирующую процессы естественного изменения ДНК и сравнивающую полученный результат с данными образца.
Аналогичная методика была применена и в отношении менее древних образцов ДНК. Насчет мезолитических образцов из работы Лазаридиса, я не читал ту часть сапплемента где описывается техническая сторона опыта. Но в другой работе упомянутого в статье Скоглунда (Skoglund et al .2012) — в неолитическах образцах результаты поссмертной гидролитической деаминации (cytosine —> thymine or guanine —> adenine) были удалены. Но у неандера разумеется из было горадо больше и пришлось придумывать методику реконструкции первоначальных нуклеотидов.Кроме того, в статье Lazardis et.al.2013 (точнее в сапплементе) содержится указание на использование урацил-ДНК-гликосилазы и эндонуклеозы при подготовке библиотек для сиквенирования.Использование этого метода значительно (!) уменьшает включение деаминированных остатков C/G→T/A (здесь подробности).
Уважаемый «любитель» Владимир Таганкин на основе большого эмпирического материала (десятки тысяч гаплотипов) провел серьезное исследование дисперсии значений локусов Y-STR. Это исследование по своему качеству превосходит многие статьи профессиональных популяционных генетиков.
В статье доктора Линча известный «феномен раздутости нефункциональной части человеческого генома» объясняется сочетанием ряда генетических факторов. Мутации, увеличивающие размер генома (дупликации), с гораздо меньшей вероятностью вредят организму, чем мутации, при которых часть генома теряется (делеции). Поэтому с увеличением частоты мутаций геном начинает непроизвольно расти. То есть причинно-следственная цепочка тут следующая:
малый размер популяции > увеличение генетического дрейфа > нарушение аккуратности репликации генома (увеличение частоты мутаций) > увеличение размера генома.
Как мне кажется, это объяснение можно применить к анализу всех мутаций, в том числе и STR (коротких тандемных потворов).
В январе и начале февраля было опубликовано несколько статей, в которых затрагивается тематика ДНК-криминалистика. Так в ходе проведенного Федеральным Бюро Расследований США аудита национальной базы данных ДНК, было обнаружено 166 ДНК-профиля, которые содержали ошибки. Часть этих ошибок появилась в результате ошибок клерков, другая часть связана с ошибками при интерпретации данных допущенных сотрудниками лабораторий. Проведенная тогда же проверка профилей ДНК в базе данных города Нью-Йорке дала аналогичные результаты. Неприятный факт обнаружения ошибок в STR-профилях ДНК поднимает старые вопрос о необходимости замены существующей системы CODIS. В более ранней работе, в которой рассматривалась роль и место устаревающей, но по-прежнему существующей системы CODIS в системе быстро развивающегося комплекса знаний о геноме человека, авторы сделали интересный вывод: несмотря на то, что маркеры CODIS часто лежат в пределах геномных и генных доменов, связанных с риском развития определенных заболеваний или отвечающих за определенные функции генома, не было найдено никаких убедительных доказательств того, что «короткие тандемные повторы», используемые в качестве маркеров CODIS, могут помочь установить физические черты человека. Наконец, в совсем новой работе по ДНК-криминалистике («Recent Advances in Forensic DNA analysis«), наряду с обсуждением сугубо технических моментов сбора и подготовки биологического материала к анализу, затрагивается и вопрос о возможных альтернативах STR (коротких тандемных повторов), т.е того типа маркеров которые лежат в основе системы CODIS. Одной из логичных альтернатив являются однонуклеотидные полиморфизмы (снипы). Одним из преимуществ снипов над STR является тот факт, что в сильнодеградированные фрагменты ДНК могут быть проанализированы только с помощью снипов. Будучи биаллельным маркером, снип может быть включен в ДНК-профиль, однако информативность одичного снипа гораздо ниже информативности STR-локусов, в силу чего процесс установления личности при работе со смесью разнородных ДНК усложняется. Хотя единчный снип менее информативен ( в силу биаллельности), чем STR, но этот недостаток можно легко избежать за счет увеличения количества SNP(снип)-маркеров, используемых при анализе. Разный уровень гетерозиготности является одной из наиболее ценных особенностей снипов. Другой положительной чертой снипов является то, что при определении снипов нет нужды на разделение сегментов по их размеру, что делает мультиплексирование и автоматизации более доступны, чем в анализе коротких тандемных повторов. Кроме того, низкая скорость мутации снипов значительно улучшает их стабильность в качестве генетических маркеров.



Для отправки комментария необходимо войти на сайт.