Две недели назад я сообщил об окончании первой фазы своего нового проекта (на первом этапе работы удалось собрать надежную выборку из более чем 5000 образцов более чем 250 различных этно-популяционных групп людей по всему миру.
Как я уже рапортовал ранее, самой сложной из запланированных на втором этапе задач являлась импутирование (импутация) отсутствующих генотипов. Читатели моего блога помнят, что две предыдущие экспериментальные попытки импутирования больших выборок — в 2013 и в 2015 — закончились неудачно (или, если говорить точнее, качество импутированных генотипов не оправдало моих завышенных ожиданий). В предыдущих опытах я задействовал мощную комбинацию программ ShapeIT и IMPUTE и метод импутирования снипов за счет использования большой референсной панели аутосомных гаплотипов (из 1000 genomes), гарантирующей более аккуратное определение генотипов.
На этот раз, я решил не повторять ошибок, и обратился к использованию других программ — в частности , к Minimac3, хорошо зарекомендовавшую себя в работе с геномами 1000G. К моему счастью, я набрел на недавно появившиеся публичные сервера, работающие с «облачным» сервисом импутирования Cloudgene. геномов.
Серверы импутирования геномов позволяют использовать полную референсную панель гаплотипов для точного определения недостающих генотипов в анализируемых данных. Пользователи подобных серверов могут загружать (предварительно фазированные или несфазированные) данные генотипов на сервер. Процедура импутирования будет осуществляться на удаленном сервере, и по окончанию этого процесса рассчитанные данные доступны пользователю для скачивания. Наряду с импутированием, подобные сервисы позволяют провести процедуру контроля качества (QC) и фазировки данных в качестве предварительного этапа процесса импутирования генотипов.
Прототипы серверов импутирования уже доступны в институте Сангера и Мичиганского университета. В дополнение к вышеназванным серверам, можно упомянуть прототип сервера поэтапной полномасштабной фазировки генотипов анализируемых образцов (прототип создан биоинформатиками Оксфордского университета). На мой взгляд, самое простое и доступное решение задачи импутирования на удаленном сервере было разработано сотрудниками Мичиганского университета. Дополнительное преимущество этому решению дает грамотная документация по использованию сервиса.
Основная рабочая лошадка сервиса — это комбинация двух или трех программ — две програмы для фазирования диплоидных генотипов в гаплоидную фазу ShapeIT и Hapi-UR , а в качестве основного ПО для самого процесса импутирования (определения) недостающих генотипов — вышеупомянутую программу Minimac3.
Описание эксперимента с импутированием генотипов на удаленном сервере
В самом начале, я разбил свою выборку на пять когорт (т.к. референсные панели на сервере также разбиты на «этнографические группы»):
- европейцы (европейцы + кавказцы) — 1715 образцов -87169 снипа
- азиаты (+американские аборигены и аборигены островов Тихого Океана) — 2356 образцов — 87044 снипа
- африканцы — 1054 образца — 86754 снипов
- палеогеномы древних жителей Евразии, Африки и Америки -340 — 594500 снипов
- смешанные группы — преимущественно мозабиты, пуэрто-риканцы и др.
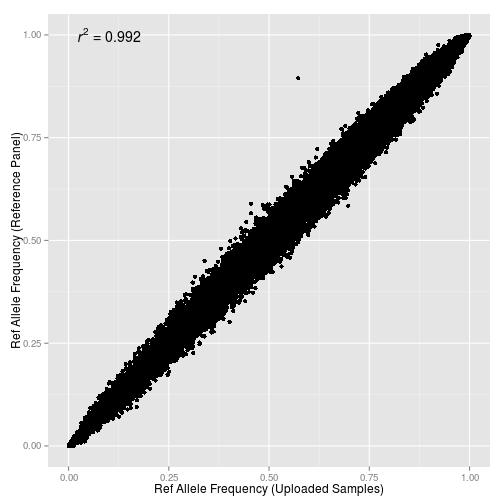
К моему вящему неудовльствию, некоторые образцы в сводной выборке не прошли контроль качества — в первую очередь это касается образцов европейцев из базы данных POPRES, а также выборок статьи Xing et al. (2010). Скорее всего, их нужно будет импутировать отдельно.
Несмотря на значительную скорость обработки генотипов на удаленном сервере, к настоящему времени эксперимент еще не доведен до конца. Пока я планирую ограничиться импутированием генотипов в 3 первых когортах (т.к. импутирование палеогеномов с помощью современных референсных панелей гаплотипов вероятнее всего приведет к искажению истинного разнообразия палеогеномов за счет проекции на современные группы населения, хотя авторы статьи Gamba et al. 2014 в сопроводительном материале к своей статье утверждают обратное).
После окончания фазирования и последующей обработки генотипов европейской когорты в программе Plink (были отсеяны все варианты с вероятностью ниже 0.9) , я получил выборку из 1715 европейцев с 25 215 169 снипами против изначальных 87169, т.е число снипов в выборке увеличилось в 290 раз!
В азиатской когорте соотношение импутированных генотипов к исходным составило чуть меньшую величину 19 048 308 / 87044 = 219.
Проверка результатов
Разумеется, все полученные результаты нуждались в дополнительной проверке качества генотипирования.
Cначала я объединил импутированную европейскую когорту с когортой палеогеномов (которая не была импутирована) и рассчитал в программе PLINK 1.9 матрицу IBS (т.е. сходства образцов в выборке между собой, эта метрика отдаленно напоминает Global Similarity в клиентских отчетах 23andme), а затем усреднил данные по популяциям и произвел по усредненным значениям иерархическую кластеризацию по признакам сходства (IBS, identity by state). Результат превзошел все мои пессимистические ожидания
Как становится очевидно из приведенной выше кластерограммы, в целом взаимное расположение популяций в кластерах соответствует (в общих чертах) взаимному географическому положению. Присутствуют, правда, и некоторые огрехи. Так, например, венгры очутились в одном кластере с русскими из Курска, норвежцы — с русскими из Смоленска, а усредненные «русские» — с американцами европейского происхождения из штата Юта и французами. Трудно сказать, в чем здесь причина, тем более что матрица была составлена по значениям IBS (идентичности по состоянию), а не IBD (идентичности по происхождению). Более подробные данные о попарных значениях IBS между популяциями выборки можно посмотреть в этой таблице
Импутированная азиатская когорта (несмотря на расширение географии за счет включения образцов коренного населения Америки и аборигенов бассейна Тихого океана) тоже оказалась на удивление надежной. Я пока не буду останавливаться на подробностях изучения этой когорты, вместо этого я размещаю здесь результаты MDS- мультдименсионального шкалирования образцов выборки, образованной в ходе слияния 2 импутированных когорт (европейской и азиатской) с 1 неимпутированной (палеогеномы). Цветовое обозначение точек соответствует определенным кластерам, выявленных в выборке с помощью алгоритма MCLUST (cледуя рекомендациям Диенека Понтикоса). Всего этих кластеров 15 и они обозначены последовательностью чисел от 1 до 15, и каждый из этих кластеров имеет свою четкую географическую привязку:
- 1 — кластер популяций ближнего Востока и Анатолии
- 2 — кластер популяций северного Кавказа
- 3 — «индоевропейский» кластер древних популяций Синташта, шнуровой культуры, Ямной культуры и т.д.
- 4 — кластер аборигенных жителей Америки (эскимосов и индейцев)
- 5 — суперкластер популяций средиземноморского и восточноевропейского региона
- 6 — сибирский кластер алтайских и самодийских популяций
- 7 — кластер популяций западной и северной Европы
- 8 — кластер палеосибирских популяций (таких как чукчи, ительмены и коряки)
- 9 — кластер аборигенных (австронезийских и тай-кадайских) популяций юго-восточной Азии (даи, атаяла и ами)
- 10 — кластер неолитических популяций
- 11 — еще один ближневосточно-средиземноморский кластер (ашкеназим, сардинцы и так далее)
- 12 — кластер североиндийских популяций
- 13 — кластер центральноазиатских популяций
- 14 — поволжские популяции
- 15 — разные групп индусов












Для отправки комментария необходимо войти на сайт.