Данная заметка представляет собой критический анализ методологических предпосылок создания неандертальского калькулятора, имплементированного в cоответствующем сервисе 23andme (Neanderthal lab). В основу заметки положен перевод технического документа 23andme (white paper), описывающего процесс создания неандертальского калькулятора.
Существует несколько методологических подходов к созданию неандертальского калькулятора (т.е инструмента для оценки того, сколько процентов ДНК в геноме анализируемого индивида имеет неандертальское происхождение). Есть несколько способов прямой экспериментальной оценки величины процента «неандертальской » ДНК с помощью ресеквенирования ДНК клиента в тех регионах, в которых ученые обнаружили возможные варианты, имеющие предполагаемое неандертальское происхождение. Но в силу технической сложности реализации этих способов и необходимости каждый раз заново производить секвенирование в полном объеме регионов неандертальского происхождения, нет особой нужды рассматривать их в этой записи. Вместо этого я предлагаю рассмотреть две оставшиеся методики определения вклада неандертальского ДНК. Хотя оба метода не без своих изъянов, они позволяют существенно снизить влияние неопределенности (ascertainment bias) в оценке вклада неандертальского ДНК, и в принципе, других приемлемых альтернатив этим методам не существует, так как в противном случае получаемый другими методами (например, D—statisticsили ABBA—BABA) разброс оценки величины неандертальского вклада будет в несколько раз отличаться от тех величин, которые получаются на выходе соответствующих программ, используемых в NationalGeographicGeno и 23andme (обе программы основаны на одном из двух нижеописанных методов).Именно по этой причине, каждая из нижеприведенных методик заслуживает отдельного рассмотрения.
- Метод PCA
На мой личный взгляд, наилучшим (как в плане аккуратности, так и в плане легкости реализации) методом оценки величины неандертальца в ДНК клиентов является метод главных компонент PCA, так как он представляет собой очень мощный инструмент для представления корреляции данных высокой размерности (порядка миллионов снипов и даже больше) в виде гораздо меньшего, некоррелирующего набора переменных, которые носят название «главные компоненты». Итак, метод главных компонент — это один из способов понижения размерности, состоящий в переходе к новому ортогональному базису, оси которого ориентированы по направлениям максимальной дисперсии набора входных данных (в нашем случае это набор генотипов снипов). Вдоль первой оси нового базиса дисперсия максимальна, вторая ось максимизирует дисперсию при условии ортогональности первой оси, и т.д., последняя ось имеет минимальную дисперсию из всех возможных. Такое преобразование позволяет понижать информацию путем отбрасывания координат, соответствующих направлениям с минимальной дисперсией. Можно отметить, что в основе метода главных компонент лежат следующие допущения: (a) допущение о том, что размерность данных может быть эффективно понижена путем линейного преобразования, и (b) допущение о том, что больше всего информации несут те направления, в которых дисперсия входных данных максимальна.
На первом этапе анализа необходимо вычислить главные компоненты отображающие дисперсию данных неандертальца по отношению данным современного человека. Для этого необходимо провести PCA анализ, в который будут включен набор снипов неандертальцев, набор снипов денисовского человека, и набор снипов шимпанзе (Clint).
Сначала скачиваем полные геномы неандертальца, денисовского человека, и шимпанзе Clint. Затем с помощью программы samtools генерируем для каждого из трех геномов файлы с геномными вариантами (vcf), отфильтровываем из полученных файлы инделы, таким образом чтобы на выходе остались только снипы и проводим аннотацию снипов с использованием базы данных dbSNP; при аннотации находятся те варианты, которые присутствуют в базе данных и им назначается соответствующий индекс, например rs4213456 (это условный пример). Затем необходимо выбрать из это файла только те cнипы, которые присутствуют в контрольной выборке с референсными популяциями современного человека. Описание примерного порядока выполнения этой задачи можно найти в двух записях в моем блоге (здесь и здесь).
В конечном итоге, по окончанию первого этапа, мы получаем три файла VCF c аннотированным снипами, которые необходимо соединить в один файл либо в vcftools, либо в Plink. Затем провести анализ PCA с двумя заданными главными компонентами (K2) в самом Plink, либо конвертировать данные в формат Eigenstrat и провести в программе Eigensoft анализ PCA (также с двумя заданными главными компонентами). Последний вариант предпочтителен, так алгоритм Eigensoftдает более точные данные за счет kernel-преобразований данных. В конечном результате проведенного анализа двух основных компонентов должны получится нормированный лист cобственных векторов — эйгенвекторов так называемый лист факторной загрузки –factor loading) для каждого из индивидуальных образцов, входящих в анализируемый набор. Первый главный компонент, PC1 , чьи значения отображаются вдоль первой оси ортогонального базиса, характеризуется максимальной дисперсией набора снипов входящих данных, эта ось отображает общее генетическое сходство архаичных людей (неандертальца и денисовского человека). Ось второго компонента , PC2 , оптимизирует дисперсию при условии ортогональности первой оси (т.е, PC1), и отображает генетическое расхождение между неандертальцами и денисовским человеком.

На следующем этапе генотипы клиентыпроецируются на плоскость, образованную двум яосями PC1 и PC2. Я полагаю, что на этом этапе в самом PCA анализе нет необходимости, вместо этого можно имплементировать метод с использованием высчитанного в первом анализе PCA листа загрузки компонентов (loadings). Подобный подход реализован, например, в программе shellfish.
В случае успешного выполнения промежуточной задачи на этом этапе, те клиенты, у которых нет неандертальского или денисовского вклада в геном, должныр авномерно распределиться в центре графика, то есть внутри условного треугольника, образованного референсными геномами неандертальца, денисовского человека и шимпанзе.В то время, как клиенты с неандертальской примесью должны будут проецироваться ближе к неандертальца .
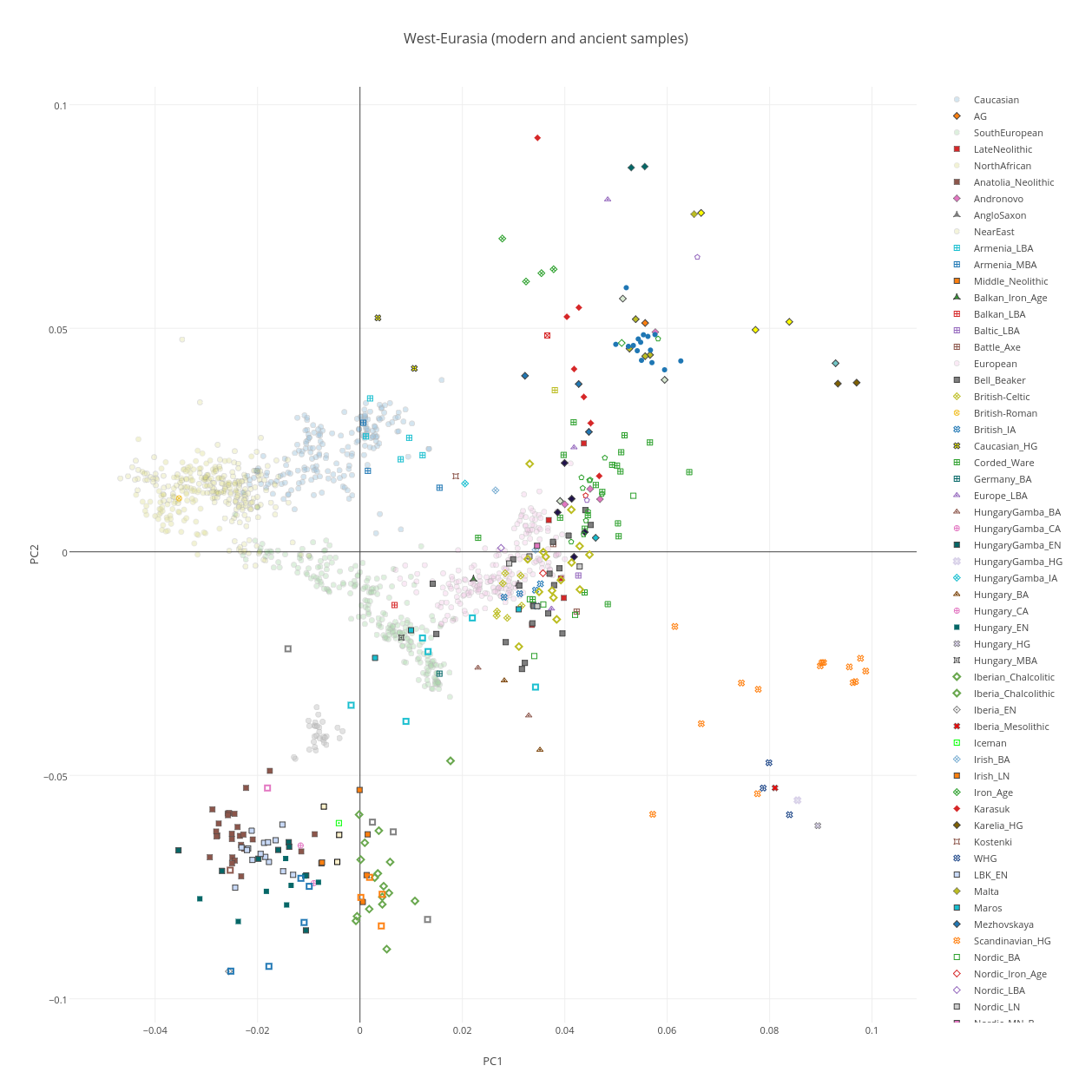
Как видно из иллюстрации к работе (Reich et al.2011), европейцы и жители Восточной Азии существенно сдвинуты в сторону неандертальцев по сравнению с афро-американцами (как видно из приведенного ниже графика, расстояние между неандертальским «углом» и положением афроамериканцеввесьма значительно, это следствие неопределенности определения предковых аллелей неандертальца по африканским популяциям, поэтому для коррекции этой дистанции в 23andme высчитали центроид генетического положения африканцев с использование данных проекта 1000G, и расчет дистанции вели от него).

На третьем этапе необходимо преобразовать PCA-координаты популяций современных людей в процент неандертальского ДНК, т.е привести к тому виду, который выдается клиенту на выходе. Для этих целей каждый клиент проецируется на расчетную «неандертальскую» ось, представляющую собой линию, соединяющий центроид предковой популяции клиента с точкой, координаты которой соответствует положению неандертальца на графике.
- Методтеговых (маркерных) снипов— NAIM (Neanderthal Ancestry Informative Markers)
Существует более прямой и простой способ вычисления неандертальского вклада в геном клиентов. Простота метода обусловлена отсутствием надобности в сравнительно сложных алгоритмах вычисления главных компонентов. Согласно известной публикации драфтовой версии генома неандертальца (Green et al., 2010), в геномах современных людей были обнаружены 13 геномных регионов, которые, как предполагают авторы, имели неандертальское происхождение. Эти регионы генома современных людей были маркированы с помощью маркерных (теговых) снипов – то есть таких снипов, в которых неандертальский вариант часто встречается в современных неафриканских популяциях людей, но отсутствует в коренных африканских популяциях.
В процитированной выше работе был предложен набор из 180 подобных снипов, которые маркируют эти 13 регионов, предположительного неандертальского происхождения. Таким образом, простым арифметическим подсчетом у современных людей количества известных неандертальских вариантов этих 180 снипов, можно было бы определить процент неандертальского вклада в геном современных людей. Ниже приведена таблица, в которых показаны физические координаты регионов-сегментов (хромосома, начало и конец сегмента – приведены в физических положениях сегмента в билде 36).

Тем не менее, несмотря на простоту метода, он характеризуется целым рядом недостатков, о которых следует упоминуть подробнее:
- Во-первых, не существует никаких формальных гарантий того, что эти варианты действительно имеют неандертальское происхождение.
- Во-вторых, даже в том идеальном случае, когда все эти 180 вариантов действительно имеют неандертальское происхождение, они охватывают только 13 геномных регионов, самый длинный из которых представляет собой сегмент длиной всего лишь в 160 000 базовых пар. Эта длина на два порядка величин ниже, чем среднестатистические 2,5% неандертальского вклада в среднестатистическом геноме современного человека неафриканского происхождения . Поэтому простой подсчет числа неандертальских вариантов в маркерных снипах, где встречается будет в 2-3 раза занижать реальный процент неандертальского вклада в клиентском геноме.
- В-третьих, существует еще несколько трудных моментов, связанных с практической реализацией этого метода.
3.1. Списка вышеупомянутых 180 снипов нет в открытом доступе, и так как в оригинальной статье было упомянуто другое количество снипов (166), похоже на то, что это число снипов варьируется в зависимости от использованного чипсета (поэтому и число снипов разное).
3.2. Технически эту проблему можно решить следующим образом. Самый простой способ состоит в определении того, какие снипы из используемого компанией чипсета попадают в эти сегменты. Например, берется первый сегмент на хромосоме 1 (начало 168 110 000 – конец 168 220 000, длина в базовых парах – 110 000) и выбираются снипы попадающие в этот регион, и так далее по всем регионам. При этом сначала надо узнать какой билд используется в контрольной выборке популяций современных людей. Если используется build 37, тогда необходимо конвертировать координаты сегментов в более ранний build 36. После того, как будут определены все снипы попадающие в эти 13 сегментов, нужно найти неандертальские варианты этих файлов (это можно сделать в базе данных неандертальских снипов) и составить список, который затем использовать в качестве затравки при сравнении с значениями снипов у современных людей.
3.3. Другой вариант более сложный, но очевидно более точный. Список снипов найденных в ходе сравнения геномов шимпанзе, 5 референсных популяций современных людей и неандертальца выгружен на сайте геномного браузера UCSC. Это большой файл (в распакованном виде 363 Mb), общее количество снипов 5 615 438. Формат файла следующий:
971 chr1 50600811 50600812 AA_AAD:0D,1A 0 + 50600811 50600812 0
971 chr1 50603655 50603656 AAD_AA:0D,2A 0 + 50603655 50603656 0
971 chr1 50604033 50604034 AADAA_:0D,1A 0 + 50604033 50604034 0
971 chr1 50605949 50605950 AAA_DA:0D,1A 0 + 50605949 50605950 0
Первая колонка представляет собой номер сегмента чтения, вторая – название хромосомы, вторая и третья – физическое положение снипа, далее идет длинная колонка с указанием характера варианта в шимпанзе, 4 популяций людей и неандертальца. «A» обозначает предковое значение аллеля, «D» — derived, т.е мутировавшее значение. После двоеточия идет специфическая неандертальская колонка (например, :0D,1A)с указанием того сколько предковых и сколько мутировавших значений снипа обнаружено в исследованных геномах неандертальцев. В данном случае, в первом снипе обнаружено 0D (0 мутировавших) и 1A (1 предковое значение). Трудность задачи состоит в определении только тех снипов, в которых у неандертальцев нет предковых значений, а встречаются только мутировавшие значения. Эти снипы — кандидаты на неандертальский вклад в человеческий геном. Затем сравнить отфильтрованный список со списком снипов в контрольной выборке (опять-таки, надо знать какой билд используется, координаты этого списока приведен по билду 36) и выбрать только те, что имеются в чипсете компании. Далее алгоритм тот же, что и выше – определяется значение снипа у неандертальца и сравнивается с соответствующим значением у современных людей. Совпадающие у неандертальца и современных людей варианты подсчитываются и определяется конечный процент неандертальского вклада.
Эксперимент.
Я решил проверить эфективность первого метода (метода PCA) на своей контрольной выборке (2778 образцов современных людей, шимпанзе, денисовского человека и неандертальского человека и 142429 снипа). В качестве рабочей программы я использовал новую версию Plink, которая позволяет использовать в анализе PCA заданные контрольные кластеры, в которые проецируются исследуемые индивиды. В качестве трех контрольных групп я выбрал, следуя рекомендациям авторов обсуждаемого исследования, геномы шимпанзе, неандертальца из Vindja и денисовского человека. Однако число априорных главных компонентов я намерено изменил, с 2 на 3 (K3), таким образом на выходе я получил эйгенвекторы трех главных компонентов. По этой причине, полученный мной график PCA несколько отличается от вышеприведенного графика 23andme (вместо PC1 и PC2 я использовал PC2 и PC3, то есть второй и третьи главные компоненты, более точно описывающие в данном случае сходство/различие геномов архаичных и современных людей).

Как видно из наших результатов, все популяции современных людей разместились внутри условного треугольника образованного дисперсией геномов денисовского человека, неандертальца и шимпанзе.
Впрочем, на графике нельзя разглядеть, какие именно популяции сдвигаются в сторону неандертальца, а какие — в сторону денисовского человека (такой сдвиг свидетельствовал бы о наличии адмикса). Чтобы устранить этот досадный артефакт графика, придется убрать с графика геномы денисовца, неандертальца и шимпанзе (из-за значительной генетической дистанции популяции современных людей сдвигаются в одну кучу).


Положение удаленных денисовца, неандертальца и шимпанзе размечено на новом графике буквенными обозначениями — D, N, Chimp. Из человеческих популяций я разметил группы африканских популяций (Africans), и коренных американцев (Native Americans). Европейские и азиатские популяций смещены в одну общую группу, с сильным креном в сторону неандертальца. Судя по всему, мои результаты, в общих чертах, практически не отличаются от результатов исследований Грина и Райха. Как отмечает Дробышевский: » «денисовские гены», несмотря на свою экзотичность, обнаружились у современных людей. Первоначально они были найдены у папуасов Новой Гвинеи и меланезийцев острова Бугенвиль (Reich et al., 2010), затем – у австралийских аборигенов (Gibbons, 2011), а полнейшее исследование вопроса констатировало наличие их у огромного числа популяций (Reich et al., 2011). Они были выявлены в тридцати трёх популяциях Океании и Юго-Восточной Азии, в том числе у папуасов Новой Гвинеи, австралийских аборигенов (даже больше, чем у папуасов), полинезицев, фиджийцев, восточных индонезийцев с разных островов, филиппинцев и у филиппинских аэта-маманва.»
Что касается неандертальца, то уже с 2010 года известно, что в целом неандертальская ДНК составляет 1-4% генома нынешних людей, живущих за пределами Африки. Авторы двух исследований, опубликованных в среду журналах Science и Nature, выяснили, что чаще всего неандертальская наследственность присутствует в нескольких генах, связанных с выработкой кератина, присутствующего в коже, волосах и ногтях. В этой части генома неандертальские аллели обнаружены у 70% европейцев и 66% азиатов.
Гораздо интереснее те мои результаты, которые отличаются от общепринятых. Так например, довольно неожиданным результатом является наблюдаемое на графике значительное смещение южноамериканских индейцев в сторону денисовского человека, причем это смещение гораздо значительнее смещения папуасов и меланезийцев, у которых были найдены «денисовские гены» в наибольшем количестве. Что это означает, трудно сказать — наличие реального сигнала смешивания в данном случае равновероятен обнаружению статистического артефакта. Впрочем, если верить работам Скоглунда этот результат может быть правдоподобным — моделирование миграций генов показало, что «денисовские» гены должны встречаться не только в Юго-Восточной Азии, но даже в некоторых группах Южной Америки (Skoglund et Jakobsson, 2011)
Оставим в стороне этот вопрос, который нуждается в более детальном изучении, и передем к расчетам процентной величины вклада неандертальских генов в популяции современных людей. Очевидно, что средняя величина этого вклада по каждой из популяций может дать только приблизительное представление о характере архаичной интрогресси неандертальских генов. Индивидуальный уровень вклада в каждой популяции может иметь большую частотную амплитуду в интервале между 1 и 6% процентами. Тем не менее, представляется возможным апроксимировать эти значения путем умножения собственного вектора (eigenvector) главных компонентов каждого индивида каждой популяции на собственное число линейного преобразования (eigenvalue), и последующим усреднением по популяции.
Ниже приведены эти усредненные значения в процентах (неандертальских генов), в порядке уменьшения. Вызывают сомнения ультра-высокие значения в первых десяти популяциях — скорее всего это результат комплексного воздействия статистических эфектов недостаточной представленности выборки, а также высокой степени гомозиготности, характерной для изолированных популяций (исландцев, албанцев и басков). Довольно высок уровень неандертальского вклада в образцах древних европейцев, хотя это и логично с точки зрения исторической модели адмикса. С другой стороны, средние значения (2-2.7%) неандертальского адмикса в популяциях Восточной Европы выглядят реалистичными. Так, например, по расчетам 23andme у меня уровень «неандертальских генов» составляет 2.67% :
| Icelandic |
10.50% |
| Norwegian |
9.00% |
| 1_Motala12 |
8.00% |
| Spain_BASC |
8.00% |
| Albanian |
7.00% |
| Korean |
7.00% |
| Tiwari |
5.11% |
| 1_LBK380 |
5.00% |
| 1_Loschbour |
5.00% |
| French_South |
4.00% |
| Kashmiri |
4.00% |
| Tubalar |
4.00% |
| Atayal_Coriell |
3.60% |
| Ami_Coriell |
3.10% |
| 1_Motala_merge |
3.00% |
| Bolivian |
3.00% |
| Croatian |
3.00% |
| Totonac |
2.80% |
| Qatari |
2.71% |
| Mixed_East_Slav |
2.57% |
| Gujarati |
2.43% |
| Ulchi |
2.39% |
| North-Russian |
2.36% |
| Center-Russian |
2.36% |
| Aonaga |
2.33% |
| British |
2.33% |
| Chenchu |
2.33% |
| East-Belarusian |
2.33% |
| Ukrainian |
2.33% |
| Finn |
2.29% |
| Latvian |
2.29% |
| Mixed_European |
2.28% |
| South-Russian |
2.27% |
| Pole |
2.26% |
| Lithuanian |
2.25% |
| West-Belarusian |
2.25% |
| Belarusian |
2.23% |
| Vepsa |
2.23% |
| Bosnian |
2.22% |
| Cree |
2.20% |
| Georgian_Imereti |
2.20% |
| Polish |
2.20% |
| Orcadian |
2.15% |
| Russian |
2.15% |
| Karelian |
2.13% |
| Welsh |
2.12% |
| Swede |
2.11% |
| Ukranians |
2.11% |
| Greek |
2.10% |
| Lithuanians |
2.10% |
| Gagauz |
2.09% |
| Croat |
2.08% |
| Slovak |
2.08% |
| Estonians |
2.08% |
| Adygei |
2.07% |
| Serb_Serbia |
2.07% |
| Toscani |
2.07% |
| French |
2.06% |
| Komi |
2.06% |
| 1_LaBrana |
2.00% |
| Algonquin |
2.00% |
| Avar |
2.00% |
| Azeri_Dagestan |
2.00% |
| Azov_Greek |
2.00% |
| Bashkir |
2.00% |
| Belgian |
2.00% |
| Bulgarians |
2.00% |
| Central-Greek |
2.00% |
| CEU |
2.00% |
| Cirkassian |
2.00% |
| Cochin_Jew |
2.00% |
| Corsican |
2.00% |
| Cretan |
2.00% |
| Croat_BH |
2.00% |
| Don_cossack |
2.00% |
| Eskimo |
2.00% |
| Haida |
2.00% |
| Hungarian |
2.00% |
| Hungarians |
2.00% |
| Inkeri |
2.00% |
| Inkeri-Finn |
2.00% |
| Italian_Abruzzo |
2.00% |
| Kets |
2.00% |
| Kosovar |
2.00% |
| Kryashen |
2.00% |
| Kuban_cossack |
2.00% |
| Lezgin |
2.00% |
| Macedonian |
2.00% |
| Meghawal |
2.00% |
| Mishar |
2.00% |
| Mixed_CEU |
2.00% |
| Mixed_East_European |
2.00% |
| Mixed_German |
2.00% |
| Mixed_Slav |
2.00% |
| Montenegrian |
2.00% |
| Mordovian |
2.00% |
| Mordovians |
2.00% |
| North_Italian |
2.00% |
| Occitan |
2.00% |
| Roma_Bulgarian |
2.00% |
| Roma_Macedonian |
2.00% |
| Romanian_Jew_2 |
2.00% |
| Russian_South |
2.00% |
| Saami |
2.00% |
| Selkup |
2.00% |
| Serb_BH |
2.00% |
| Slovenian |
2.00% |
| South_Greek |
2.00% |
| Swedish |
2.00% |
| Tabassaran |
2.00% |
| Tatar_Lithuanian |
2.00% |
| Velama |
2.00% |
| West_Greenland |
2.00% |
| French_Basque |
1.95% |
| Chechens |
1.94% |
| Iberian |
1.94% |
| Chuvash |
1.94% |
| Tatar |
1.93% |
| Balkars |
1.92% |
| German |
1.92% |
| North-Ossetian |
1.92% |
| Hant |
1.89% |
| North_Greek |
1.89% |
| Georgians |
1.88% |
| Lak |
1.88% |
| Abhkasians |
1.85% |
| Sardinian |
1.84% |
| Udmurd |
1.84% |
| Maris |
1.82% |
| Romanians |
1.82% |
| Georgian_Laz |
1.80% |
| Kumyks |
1.80% |
| Lodi |
1.80% |
| Mansi |
1.77% |
| Chukchis |
1.75% |
| Crimean_Tatar |
1.75% |
| Italian_Piedmont |
1.75% |
| Ket |
1.75% |
| Moldavian |
1.75% |
| Vaish |
1.75% |
| Hallaki |
1.67% |
| Lezgins |
1.67% |
| Ossetian |
1.67% |
| Tlingit |
1.67% |
| Greek-Islands |
1.63% |
| Turks |
1.63% |
| Armenians |
1.60% |
| Nogais |
1.60% |
| Selkups |
1.60% |
| Hakas |
1.57% |
| Ashkenazy_Jews |
1.56% |
| Apache |
1.50% |
| Jew_Tat |
1.50% |
| Kabardin |
1.50% |
| Karitiana |
1.50% |
| Kurds |
1.50% |
| Nenets |
1.50% |
| Samaritians |
1.50% |
| Santhal |
1.50% |
| Srivastava |
1.50% |
| Syrian_Jew |
1.50% |
| Tuva |
1.50% |
| Uygur |
1.50% |
| Mexican |
1.45% |
| Italian_Jew |
1.40% |
| Portugese |
1.40% |
| Tajiks |
1.40% |
| Kyrgyzians |
1.38% |
| Roma_Slovenian |
1.38% |
| Altaians |
1.36% |
| Koryaks |
1.33% |
| Pashtun |
1.33% |
| Satnami |
1.33% |
| Sicilian |
1.33% |
| Yakut |
1.31% |
| Cypriots |
1.30% |
| Spaniards |
1.30% |
| Turkmen |
1.30% |
| French_Jew |
1.29% |
| Iraqi_Jews |
1.29% |
| Sephardic_Jews |
1.29% |
| Turkmens |
1.29% |
| Parsi |
1.28% |
| Buryats |
1.27% |
| Pathan |
1.27% |
| Tadjik |
1.27% |
| Athabask |
1.25% |
| Iran_Jew |
1.25% |
| Kurd_Jew |
1.25% |
| Nganassans |
1.25% |
| Nysha |
1.25% |
| Azeri |
1.22% |
| Mixtec |
1.22% |
| Tharu |
1.20% |
| Tunisian_Jew |
1.20% |
| Uzbek |
1.20% |
| Evenkis |
1.18% |
| Kazakhs |
1.18% |
| Roma |
1.17% |
| Tuvinians |
1.17% |
| Druze |
1.16% |
| Karakalpak |
1.14% |
| Mongolians |
1.14% |
| Uzbeks |
1.13% |
| Ojibwa |
1.10% |
| Buryat |
1.00% |
| Cochimi |
1.00% |
| Cucupa |
1.00% |
| Dolgan |
1.00% |
| Dolgans |
1.00% |
| Even |
1.00% |
| Evenk |
1.00% |
| Hazara |
1.00% |
| Huichol |
1.00% |
| Kalash |
1.00% |
| Kalmyk |
1.00% |
| Kamsali |
1.00% |
| Koryak |
1.00% |
| Kumiai |
1.00% |
| Lambadi |
1.00% |
| Luiseno |
1.00% |
| Maya |
1.00% |
| Mongol_Halha |
1.00% |
| Nganassan |
1.00% |
| Oroqen |
1.00% |
| Pima |
1.00% |
| Roma_BH |
1.00% |
| Romanian_Jew_1 |
1.00% |
| Romanian_Jew_3 |
1.00% |
| Shor |
1.00% |
| Surui |
1.00% |
| Tharus |
1.00% |
| Tsimsian |
1.00% |
| Uyghur |
1.00% |
| Uzbekistan_Jew |
1.00% |
| Uzbekistani_Jews |
1.00% |
| Vysya |
1.00% |
| Yukaghirs |
1.00% |
| Sindhi |
0.91% |
| Hezhen |
0.86% |
| Xibo |
0.80% |
| Navajo |
0.78% |
| Bhil |
0.75% |
| Brahmins_UP |
0.75% |
| Burusho |
0.75% |
| Mongola |
0.75% |
| Naga |
0.75% |
| Iranians |
0.71% |
| Daur |
0.67% |
| Kshatriya |
0.67% |
| Mala |
0.67% |
| Moroccan_Jews |
0.67% |
| Japanese |
0.58% |
| Chinese_Dai |
0.53% |
| Evens |
0.50% |
| Kol |
0.50% |
| Morocco_Jew |
0.50% |
| Mumbai_Jews |
0.50% |
| Scheduled_Caste_UP |
0.50% |
| South_Han |
0.50% |
| Tu |
0.50% |
| North_Han |
0.45% |
| Brahui |
0.45% |
| She |
0.44% |
| Tujia |
0.44% |
| Iraki |
0.43% |
| Naxi |
0.43% |
| Dharkars |
0.40% |
| Han |
0.40% |
| Kanjars |
0.40% |
| Miaozu |
0.40% |
| Velamas |
0.38% |
| Balochi |
0.33% |
| Chenchus |
0.33% |
| Dusadh |
0.33% |
| Hakkipikki |
0.33% |
| Lahu |
0.33% |
| Piramalai_Kallars |
0.33% |
| Yizu |
0.33% |
| Colombian |
0.25% |
| Chamar |
0.22% |
| Syrians |
0.22% |
| Dai |
0.20% |
| Libyan_Jew |
0.17% |
| Makrani |
0.08% |



































{kind=link}
Для отправки комментария необходимо войти на сайт.