Компания 23andme не нуждается в особом представлении читателям этого блога. Вплоть до конца прошлого года компанию занимало существенный сегмент рынка персональной геномики, ориентированного на предоставление клиентам информации о генетических медицинских рисках (genetic risks) и генетической генеалогии (genetic origin). Информация о медико-генетических рисках содержалась в ряде сервисов портала компании, а также в доступном для скачивания отчета о генетических рисках и, разумеется, в первичных данных генетического отчета, в котором содержались значимые с точки зреемя медико-генетического диагностирования генетические полиморфизмы (SNP).
Всвязи с известными событиями и последующим за ними предписанием USA Food and Drug Administration (FDA) компании 23andme о запрете выпуска на рынок услуг персонального геномического диагностирования своего «медицинского девайза» (т.е интерпретации медико-генетических рисков развития заболеваний), компании пришлось сузить свою сферу деятельности до оказания генетико-генеалогических услуг.
Несмотря на это досадное обстоятельство, сказавшееся нелучшим образом на динамике увеличения клиентской базы компании, нужно помнить, что все клиенты сохранили доступ к своим первичным данным тестирования (т.е списку снипов с генотипами). И при вдумчивом, творческом подходе любой человек может не только «вытащить» из этих «cырых данных» важную с точки зрения медицины информацию, но и заменить спомощью полученной информацией результаты более традиционных тестов.
Каковы могут быть варианты использования данных 23andmе не в привычных генеалогических целях, а скажем для получения сведений, который могут впоследствии пригодится для молекулярного диагностирования?
Я приведу пару примеров такого использования.
Определение HLA-фенотипа.
На мембране клеток организма присутствуют продукты генов всех локусов, размещенных на обеих нитях 6-й хромосомы.
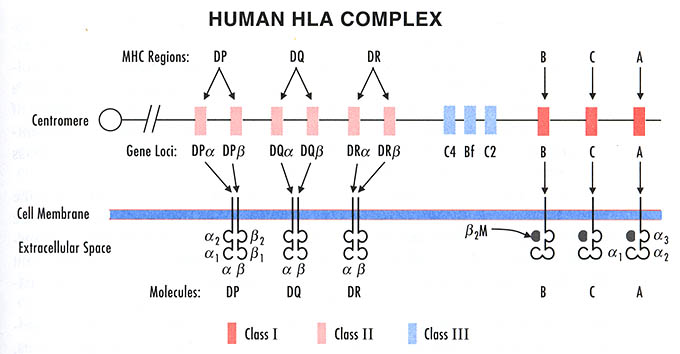
Это означает, что HLA-гены наследуются по кодоминантному типу, т. е. одну хромосому ребенок наследует от матери, а другую – от отца. Как уже упоминалось, совокупность генов, расположенных на одной хромосоме, составляет гаплотип. Таким образом, у человека два гаплотипа и каждая клетка организма несет на себе диплоидный набор антигенов системы HLA, один из которых кодируется HLA-генами матери, а другой – отца. Исключение составляют половые клетки (яйцеклетка и сперматозоид), каждая из которых содержит в своем ядре только по одному гаплотипу.
Антигены гистосовместимости, выявляемые на клетках конкретного человека, составляют HLA-фенотип. Для его определения необходимо произвести фенотипирование клеток индивида. Как правило, “типируются” лимфоциты периферической крови. До настоящего времени в большинстве лабораторий HLA-A. В, С и DR-антигены определяют при помощи серологических методов, в частности, лимфоцитотоксического теста. тот тест основан на способности анти-НLА-антител в присутствии комплемента разрушать лимфоциты, несущие соответствующие антигенные детерминанты. Гибель клеток демонстрируется при помощи добавления трипанового синего. При этом мертвые поврежденные клетки окрашиваются, и под микроскопом учитывается их количество.
Эти тесты часто требуются в ходе стандартных медицинских процедур обследования во время начала беременности, или для изучения этологии аутоимунных заболеваний. Еще более важно определение гистосовеместимости в транплантологии, где типирование HLA-фенотипа донора является обязательным условием.
Однако, с приходом новых микроматричных технологий опеределния нуклеотидов ДНК и биоинформатических методов рутинной обработки последовательности человеческих геномов , появился дешевая и относительно простая альтернатива классическим серологическим тестам (которые стоят в интервале от 100 до 500 долларов).
Я не буду останавливаться на принципиальном описании процедур, с помощью которых на основании данных 23andme можно с помощью метода «импутирования» определить HLA-фенотип, так как в прошлом году я уже разместил в этом блоге пошаговую инструкцию для выполнения этой задачи.
Впрочем, уже после того, как я отписался на эту тему здесь, в департаменте биостатистики Университета Вашингтона был разработан алгоритм HIBAG который принципиально мало чем отличается от алгоритма HLA*IMP (в обеих алгоритмах используется training model, позволяющая определять фенотип HLA по снипам 23andme). Входные данные программного решения этого алгоритма (язык R) представляют собой формат Plink. А так как в последней версии Plink была включена нативная поддержка формата 23andme, то преобразовать данные 23andme в бинарный формат Plink не сооставит особого труда. Что касается обработки данных в HIBAG, то примерный порядок выполнения команд выглядит следующим образом:
# Load the published parameter estimates from European ancestry
model.list <- get(load(«European-HLA4.RData»))#########################################################################
# Import your PLINK BED file
#
yourgeno <- hlaBED2Geno(bed.fn=».bed», fam.fn=».fam», bim.fn=».bim»)
summary(yourgeno)
# HLA imputation at HLA-A
hla.id <- «A»
model <- hlaModelFromObj(model.list[[hla.id]])
summary(model)
# HLA allele frequencies
cbind(frequency = model$hla.freq)
# SNPs in the model
head(model$snp.id)
# «rs2523442» «rs9257863» «rs2107191» «rs4713226» «rs1362076» «rs7751705»
head(model$snp.position)
# 29525796 29533563 29542274 29542393 29549148 29549597
# best-guess genotypes and all posterior probabilities
pred.guess <- predict(model, yourgeno, type=»response+prob»)
summary(pred.guess)
pred.guess$value
pred.guess$postprob
Панель метилирования Яско
В последние 10 лет, крупные генетические исследования выявили сотни генных мутаций, которые возникают чаще у аутичных пациентов, чем в общей популяции. Тем не менее, каждый пациент имеет только одну или несколько из этих мутаций, что затрудняет разработку лекарств против болезни. В настоящее время, изучением генетических факторов аутизма занимается большое количество врачей-генетиков, одним из них является доктор Эми Яско занимается исследованиями генных мутаций у аутистов. Как показали многочисленные молекулярно-генетические обследования и спектрометрия аминокислот, органических кислот и карнитинов, значительное количество аутистов страдает метаболическими нарушениями. Есть виды аутизма, вызываемые именно этими генетическими нарушениями обмена вещест.
Доктор Эми Яско разработала тест на панель метиляции Яско — тест этот дорогой, стоит 500 долларов, в этой проверяют что-то около 30 генных полиморфизмов (снипов). Выбор снипов в этой панели мотивирован тем, что эти снипы связаны с определенными генами на «молекулярно-биохимическом пути метиляции» (methyliation pathway), т.е генами которые влияют на способность организма выполнять ряд ключевых биохимических функций. Наличие генетических дисбалансов, т.е снипов в пути метиляции, будет ограничивать эффективность пути метиляции.
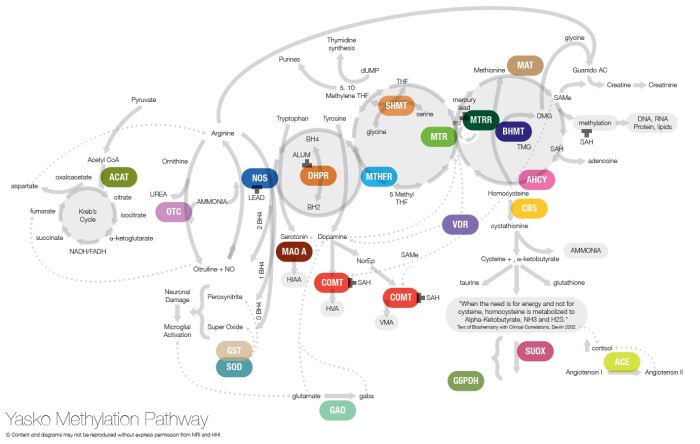
К счастью клиентов 23andme, чипсет снипов этой компании включает в себя если не все, то большую часть снипов панели Яско.
Один из проектов, возникший всвязи с неудовлетворенной потребностью клиентов в более развернутой и детальной обработке данных 23andme, Genetic Genie предлагает условно-бесплатный сервис с помощью которого данные релевантных снипов можно привести к традиционному виду таблицы с отчетом по панели Яско:
| Gene & Variation |
rsID |
Alleles |
Result |
| COMT V158M |
rs4680 |
AA |
+/+ |
| COMT H62H |
rs4633 |
TT |
+/+ |
| COMT P199P |
rs769224 |
GG |
-/- |
| VDR Bsm |
rs1544410 |
CC |
-/- |
| VDR Taq |
rs731236 |
__ |
no call |
| MAO-A R297R |
rs6323 |
TT |
+/+ |
| ACAT1-02 |
rs3741049 |
AG |
+/- |
| MTHFR C677T |
rs1801133 |
GG |
-/- |
| MTHFR 03 P39P |
rs2066470 |
AG |
+/- |
| MTHFR A1298C |
rs1801131 |
GG |
+/+ |
| MTR A2756G |
rs1805087 |
AA |
-/- |
| MTRR A66G |
rs1801394 |
GG |
+/+ |
| MTRR H595Y |
rs10380 |
CC |
-/- |
| MTRR K350A |
rs162036 |
AA |
-/- |
| MTRR R415T |
rs2287780 |
CC |
-/- |
| MTRR A664A |
rs1802059 |
AG |
+/- |
| BHMT-02 |
rs567754 |
CC |
-/- |
| BHMT-04 |
rs617219 |
AA |
-/- |
| BHMT-08 |
rs651852 |
__ |
no call |
| AHCY-01 |
rs819147 |
__ |
no call |
| AHCY-02 |
rs819134 |
__ |
no call |
| AHCY-19 |
rs819171 |
__ |
no call |
| CBS C699T |
rs234706 |
GG |
-/- |
| CBS A360A |
rs1801181 |
__ |
no call |
| CBS N212N |
rs2298758 |
__ |
no call |
| SHMT1 C1420T |
rs1979277 |
__ |
no call |
Несмотря на то, что на выходе клиент получает готовый частный отчет по тесту Яско, медико-биологическая интерпретация результатов не так уж и проста, и требует определенной интеллектуальной сноровки и общегенетической эрудиции в плане понимания того, какую функцию выполняет тот или иной ген. Строго говоря, при грамотной интерпретации этих результатов, можно самостоятельно составить себе диету из витаминов-пищевых добавок, которые позволяет компенсировать обусловленный генетическим дисбалансом дефицит тех или иных энзимов.Примерный образец интерпретации можно посмотреть здесь














Для отправки комментария необходимо войти на сайт.