Еще когда появились первые анонсы препринта статьи Haak et al. 2015, можно было сделать интуитивные предположения о том, что использованные в работе образцы палеогеномов будут всесторонне изучены не только авторами статьи, но и многочисленными любителями, причем ожидаемая степень детализации полученной картины генетического разнообразия будет предположительно выше именно у последних (т.е всевозможных геномнных блоггеров).
Так оно и вышло. Давид Веселовский из Eurogenes провел целый ряд экспериментов с объединенным базовым набром «геномов» современных популяций и так называемых древних геномов. В частности, в одном из своих анализов он задействовал новую программу qpAdm из последней версии пакета Admixtools, и в ходе пробного моделирования геномов представителей ямной культуры из самарской культуры был наилучшая аппроксимация (fit, подгонка) была получена в комбинации 51.4% генома охотников-собирателей Самары и 48.6 современных грузин (STD 0,032, chisq 3,890, р-value 2.20661e-22). Образцы палеогеномов представителей шнуровой керамики могут быть в свою очередь смоделированы как 73% геномов ямников + 27% палеогеномов Esperstedt_MN (STD 0,060, chisq 2,621, р-value 9.74968e-06).
Это интересный результат, главным образом потому данные лингвистики позволяют предположить, что ранние индоевропейцы — по-видимому, кочевники ямной культуры или их предки — были в тесном контакте с прото-картвельскими популяциями. Похожий результат был получен авторами статьи (у которых представители ямной культуры выступали как 50% -50% смесь геномов карельских охотников-собирателей и армян), а также в моих экспериментах, в которых геномы современных белорусов были представлены гибридной моделью современных геномов армян и палегеномов шведских охотников-собирателей Motala.
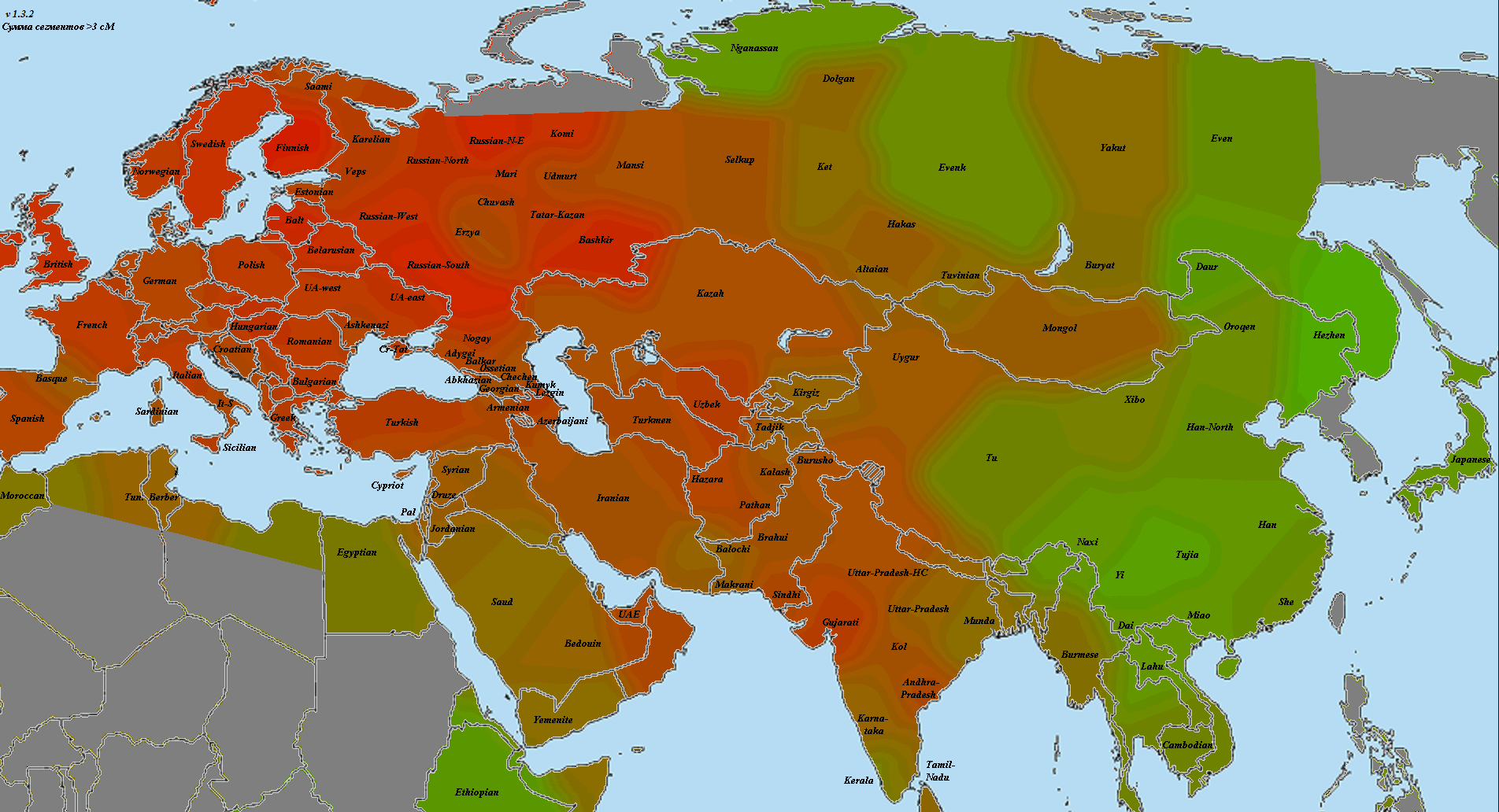
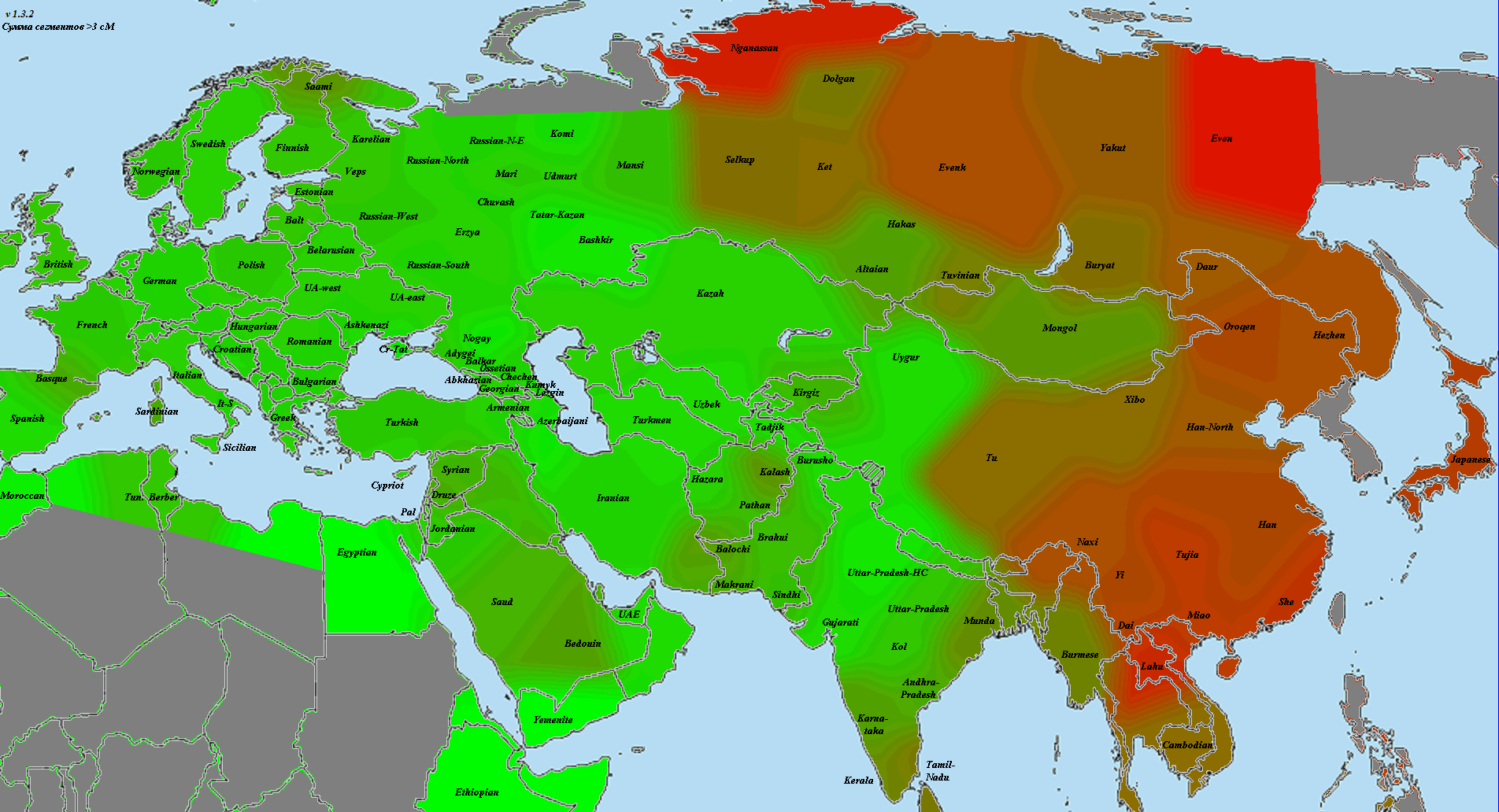
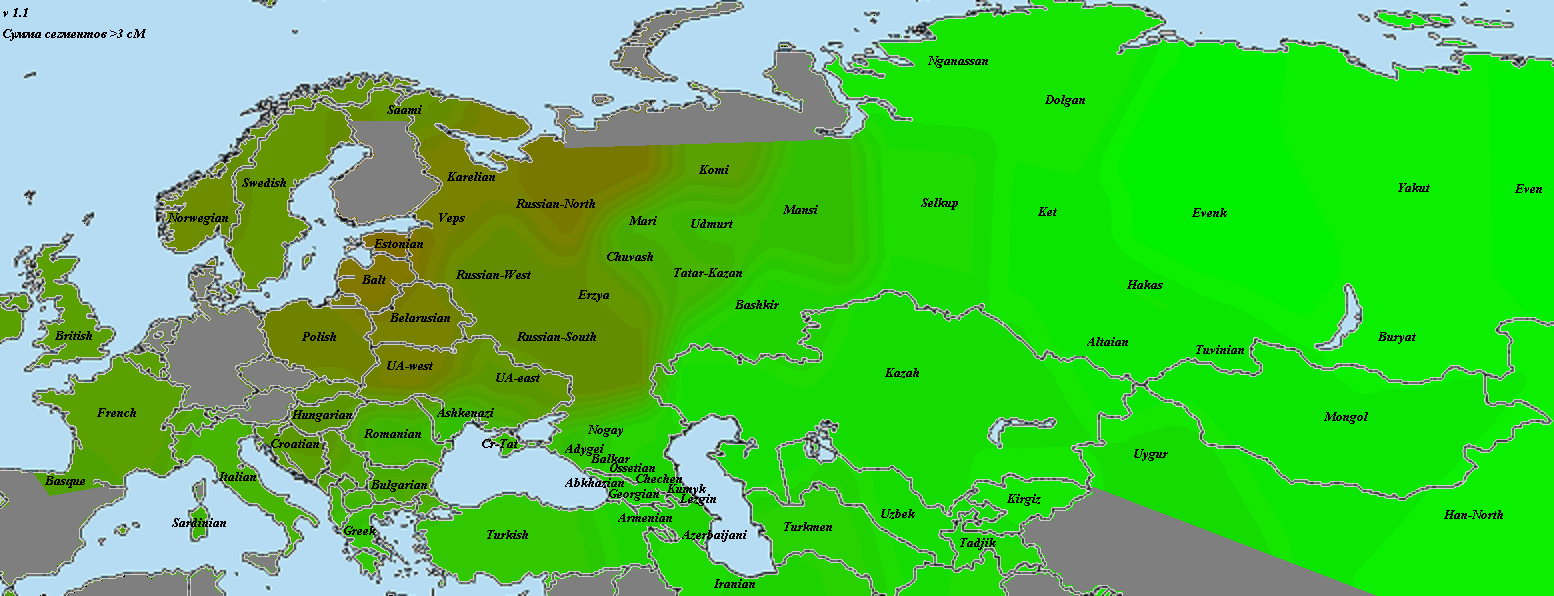
Впрочем, я согласен с Веселовским — главная проблема с подобными ретроспективными анализами заключается в том, что про причине отсутствия большого количества достоверных древних палеогеномов, популяционные генетики часто вынуждены моделировать древние популяции посредством комбинаций современных популяций. Как отмечает Веселовский, в генофонде современных грузин присутствует (по его оценке) 20% так называемого ANE-компонента, который, вероятно, прибыл на Кавказ из Евразийской степи. Если это так, то алгоритм qpAdm может переоценить «кавказский» компонент в геномах ямников, по крайней мере, на 10%.
В другом своем анализе Веселовский уделил особое внимание проблеме происхождения одного из основных компонентов в геноме древних ямников. Так например, анализ Admixture в Haak et al. 2015 включает в себя ряд интригующих компонентов с К = 16 до К = 20, которые, как правило составляют более 40% от генетической структуры потенциально прото-индо-европейских геномов ямников. Веселовский выделил компонент сигнализирующий этот тип «адмикса» и подробно изучил его. Заслуживает внимание тот факт, что компонент достигает своего пика на Кавказе и в горах Гиндукуша, и в целом показывает сильную корреляцию с регионами относительно высокой частоты связанных с палеогеномом MA1 компонентами происхождения (ANE). С другой стороны, другой компонент ямников достигает пиковых значений у ранних европейских фермеров (EEF), у которых отсутствует компоент ANE.
Выделенные Веселовским 3 основные компоненты-составляющие геномов ямников были преобразованы в синтетические популяции (центрально-азиатская, европейская и неолитическая европейская), которые в свою очередь использовались в качестве подмножества для вычисления векторов загрузки (loadings) в PCA анализе полного набора современных популяций.
https://drive.google.com/file/d/0B9o3EYTdM8lQak82NFVYSUJfWGc/preview
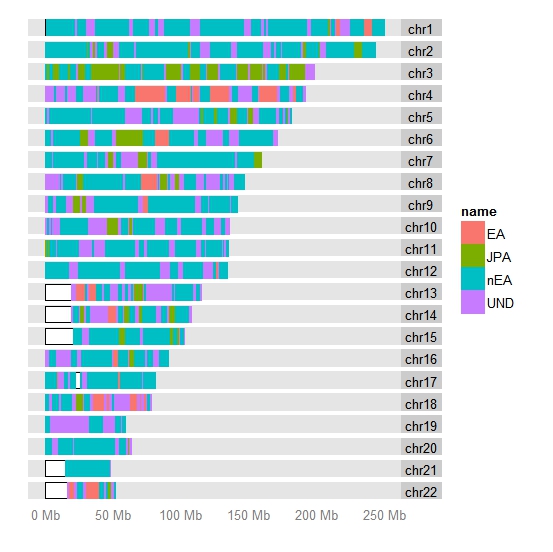
Очевидно, более детальный расклад и анализ вклада различных компонентов геномов палеоевропейцев в геном современных жителей Европы можно найти в подробном анализе Сергея Козлова «Палеоевропейцы из работы Haak et al, 2015 в свете анализа на IBD-сегменты«.
Как я уже упоминал ранее, мой опыт с «выведением» предкового аутосомного компонента индоевропейцев (обозначенного в статье Lazaridis et al. 2013 сокращением ANE) полностью удался. Поскольку всем очевидно, что этот компонент родственен «североиндийскому предковому компоненту» (ANI — обозначение из статьи Reich et al. 2009 и Moorjani et al 2011) о структуре генофонда индийских этнических групп), я взял 10 индийских этнических групп, имеющихся в кураторском наборе лаборатории Райха и проанализировал эту выборку в Admixture на пропорции вхождения их геномов в 2 априорно заданные кластеры. Первый кластер ANE был априорно задан 40 синтетическим индивидами, сгенерированными в программе Plink на основании расчитанных ранее частот аллелей «чистого» компонента ANE. В качестве дополнительного контрольного образца я использовал геном Malta1, т.к. он содержит в себе наивысшее содержание компонента ANE. Второй кластер был задан 4 индивидами Onge (одна из аборигенных народностей Андаманских островов). Как неоднократно указывалось в литературе, именно жители Андаманских островов являются самыми «чистыми» носителями т.н «южно-индийского» предкового компонента ASI (на континенте чистых носителей этого «компонента» не осталось, в том числе и среди популяций дравидов, ведда и мунда). После нескольких экспериментов по эвристическому методу проб и ошибок, я получил более или менее приемлимое разделение индивидов на 2 кластера, а затем вычислил частоты аллелей в каждом из этих кластеров. Любопытно, что в ходе опыта, удалось не только выделить компонент ANI, но и добиться неплохого уровня дискримнации между компонентом ANI, ANE, и благодаря этому, оба компонента могут быть включены в мой следующий этно-популяционный калькулятор.
Надежность компонентов я проверил на собственных данных. В рабочей модели калькулятора K14 удельное распределение этно-генографических компонентов моего генома выглядит следующим образом:
68.75% — европейский мезолитический компонент
13.12% — северо-евразийский компонент ANE
10.23% — европейский неолитический компонент
4% — ANI (северо-индийский предковый компонент)
1.6% — кавказский компонент
1.2% — алтайский компонент
0.2% — сибирский компонент
Затем я использовал 120 древних образцов аутосомной ДНК человека (начиная с верхнего палеолита до бронзового и железного веков) из последней работы и проработал их в бета-версии своего этно-популяционного калькулятора K14. Я надеялся выделить компонент ANE из ANI, но из таблицы видно, что это фактически один и тот же компонент
Когда я закончу полномерную импутацию всего набора данных от лаборатории Райха, я займусь проведением аналогичных экспериментов. А пока — примерно месяц назад я сообщил о начале первого этапа своего нового проекта. Согласно первоначальному замыслу, на первый этап — фазирование и импутация данных выборок из статей Haak et al .2015 (preprint) и Lazaridis et al. 2014 — я отводил месяц. Так оно и получилось.
В качестве затравки для импутирования я использовал набор 424329 снипов на 22 аутосомных хромосамх. Набор состоял из снипов, прошедших стандратный геномный контроль качества. Фазирование и импутация снипов я проводил с помощью пайплайна Molgenis.
По окончанию этого вычислительно-емкого процесса, мною был получен набор из примерно 5 миллионов снипов; после отсева не входящих в панели Illumina снипов у меня осталось 913841 снипов.
Ниже приведена похромосомная статистика снипов до и после импутации данных.
Как видно, на всех хромосомах (за исключением 19 и 20) количество снипов увеличилось примерно в два раза.
Для оценки качества импутации я сравнил импутированные генотипы своих данных с известными данными из своих сырых данных (снипы с иллюминовского чипсета 23andme) на предмет конкорданса (соответствия).
Оказалось, что у 6.5% импутированных генотипов оба варианта не совпадали с генотипам в rawdata от 23andme, у 17.33% — не совпадал один из двух вариантов. Таким образом, качество импутации составляет примерно 76.18%, что неплохо, учитывая что среднее значение качества импутации в программе IMPUTE v2 + SHAPEIT составляет примерно 69%.
 Chromosome Pre-imputation Post-imputation Percentage of imputed snps
Chromosome Pre-imputation Post-imputation Percentage of imputed snps
1 36638 88155 41.56
2 40140 90003 44.60
3 33218 62030 53.55
4 23594 54462 43.32
5 19731 55284 35.69
6 27979 56485 49.53
7 22804 49172 46.38
8 23072 48756 47.32
9 19369 42438 45.64
10 25340 49666 51.02
11 23145 46434 49.84
12 16967 45668 37.15
13 14998 35626 42.10
14 15529 36429 42.63
15 14663 27844 52.66
16 15034 33806 44.47
17 7799 24949 31.26
18 11697 27709 42.21
19 7102 17715 40.09
20 12654 5054 -39.94
21 6495 2572 -39.60
22 6361 13584 46.83
424329 913841 36.74
Для проверки полезности полученного набора (объединенного набора «реальных» и импутированных снипов), я соединил его с 112 образцами человеческих палеогеномов из новой статьи Haak et al. 2015. Полученный таким образом набор я проанализировал методом выделения главных компонент, первые две из которых я впоследствии использовал для построения графика главных компонент. Как мне кажется, получилось красиво и правдоподобно.

Через неделю работы в GoogleCloud, получил результаты второго цикла обработки (импутации и фазировки) палеогеномов. Напомню, задачей ставилось увеличение числа снипов палеогеномов до уровня, позволяющего проводить исследования с привлечением сторонних данных по современным человеческим популяциям (т.е не только по тем популяциям, которые включены в кураторский набор лаборатории Рейха, но и другим наборам, генотипированным на платформе Illumina; и что самое главное — с привлечением данных конкретных пользователей 23andme и FTDNA).
И если результатами первой части я был вполне доволен, то этого нельзя сказать о второй части. Теперь я понимаю, что ошибка содержалась в самом дизайне цикла второй части, в которой для импутации и фазирования использовались только реальные и «симуляционные» палеогеномы. В результате, хотя импутация и улучшила взаимное позиционирование палеогеномов в пространстве главных компонент генетического разнообразия, однако при слиянии импутированного в автономном режиме набора палеогеномов с набор полученным в первой части проекта, получилась картина. в которой палеогеномы образуют как бы параллельную субструктуру по отношению к современным популяциям.
Данное обстоятельство объясняется тем, что у древних геномов людей больше общего разнообразия между собой, чем с геномами современных людей (у которых в результате многочисленных генетических дрейфов и бутылочных горлышек большая часть разнообразия была потеряна). По этому причине, при независимой импутации древних геномов их сходство между собой только усилилось, а дистанция с современными популяциями увеличилась. Примечательно при этом, что пропорции вилкообразного разделения генетического разнообразия такие же, как и у современных людей.
На графике PCA эта ситуация прослеживается особенно хорошо, где отчетиливо видно наложение этих двух V-вилок друг на друга (см. нижний график)
Это означает одно — работу над проектом надо продолжить


















































{kind=link}
Для отправки комментария необходимо войти на сайт.