Ранее я уже отрапортовал о создании двух новых моделей для стандартного этно-популяционного калькулятора, в разработке которых использовались геномы людей, cамостоятельно указавшими свое происхождение (self-reported ancestry).
К сожалению, очень часто субъективная оценка собственного происхождения (указываемого респондентами в опросниках) недостаточно надежна для статистических методов анализа происхождения, поскольку некоторые люди либо сообщают ложные сведения о своей родословной или же просто не знают о своем истинном происхождении. Что еще хуже, — во многих публичных популяционных выборках мы не находим никаких сведений о точном этническом составе людей в выборке . Как многие из вас знают, существует множество способов достаточно точной оценки происхождения индивида на основе данных SNP генотипирования.
Самый простой способ сводится к следующему: сначала исследователь объединяет генотипы из своего исследования с генотипами образцов в референсной панели (например:
HapMapили 1000 геномов), затем находит пересечение SNP-ов в каждом наборе данных, а затем запускает программу кластеризации, чтобы увидеть, каким образом образцы исследования группируются с популяциями референсных панелей. В принципе, сам процесс несложный, но требует немало времени
К счастью, в 2014 году лабораторией Alkes была предложена программа которая, по сути, значительно облегчает процесс, выполняя большую часть работу за вас. Программа называется SNPWEIGHTS и можно скачать здесь. Говоря простым языком, программа принимает в качестве входных данных генотипы SNP-ов, самостоятельно находит пересечение генотипов SNP с генотипами в эталонной выборке , рассчитывает веса SNP-ов на основе предварительно настроенных параметров, чтобы построить первую пару главных компонентов (иначе говоря, cобственных векторов), а затем вычисляет процентное значение происхождения индивидуума из каждой предковой популяции (кластера).
Для того, чтобы запустить программу, необходимо убедится в том, что в вашей системе установлен Python, и что ваши данные генотипирования приведены в формате EIGENSTRAT. Краткую инструкции по преобразованию в формат EIGENSTRAT с помощью инструмента convertf можно почитать здесь. Данные аутосомного генотипирования FTDNA или 23andme можно напрямую преобразовать в формат EIGENSTRAT с помощью утилиты aconv от Феликса Чандракумара (либо любого самописного софта).
Затем необходимо загрузить сам пакет SNPWEIGHTS и референтную панель с весами снипов.
- Панель весов SNP для популяций Европы и Западной Африки можно скачать здесь.
- SNP веса для населения Европы, Западной Африки и Восточной Азии можно скачать здесь.
- SNP веса для населения Европы, Западной Африки, Восточной Азии и популяций американских индейцев можно скачать здесь.
- SNP веса для популяций северо-западной, юго-восточной части Европы, ашкеназских евреев и можно скачать здесь.
Затем необходимо создать файл параметров par.SNPWEIGHTS с названиями входных файлов EIGENSTRAT, референтной панели, и файл c результатами. Например:
input_geno: data.geno
input_snp: data.snp
input_ind: data.ind
input_pop: CO
output: ancestry.txt
И, наконец, нужно запустиь программу с помощью команды inferancestry.py —par par.SNPWEIGHTS. Для того чтобы программа работала, убедитесь, что inferancestry.info и файл референтной панели находятся в том же каталоге, что и файл inferancestry.py.
Полученные результаты можно использовать для разных целей. Например, можно сгенерировать два информативные графика.
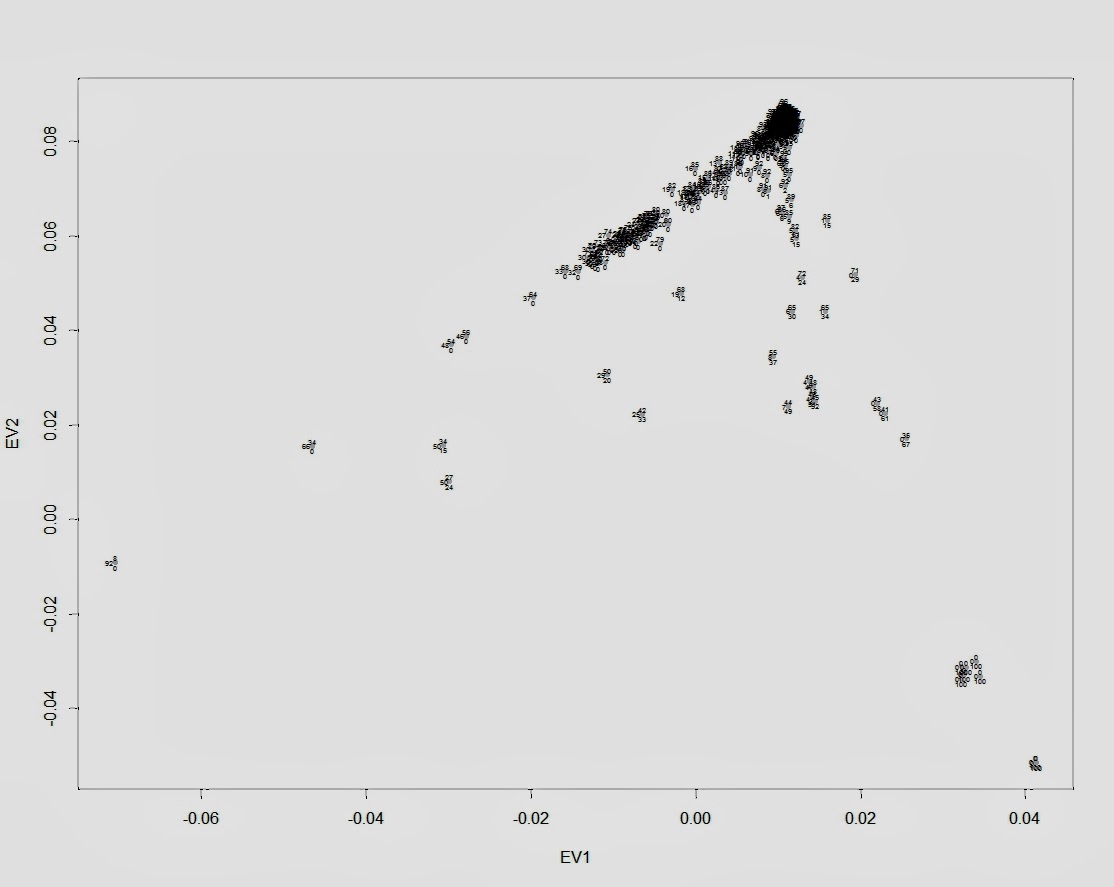
Первый график — обычный график PCA c двумя первыми компонентами (собственными векторами) и наложенный на график процентный расклад компонентов происхождения:
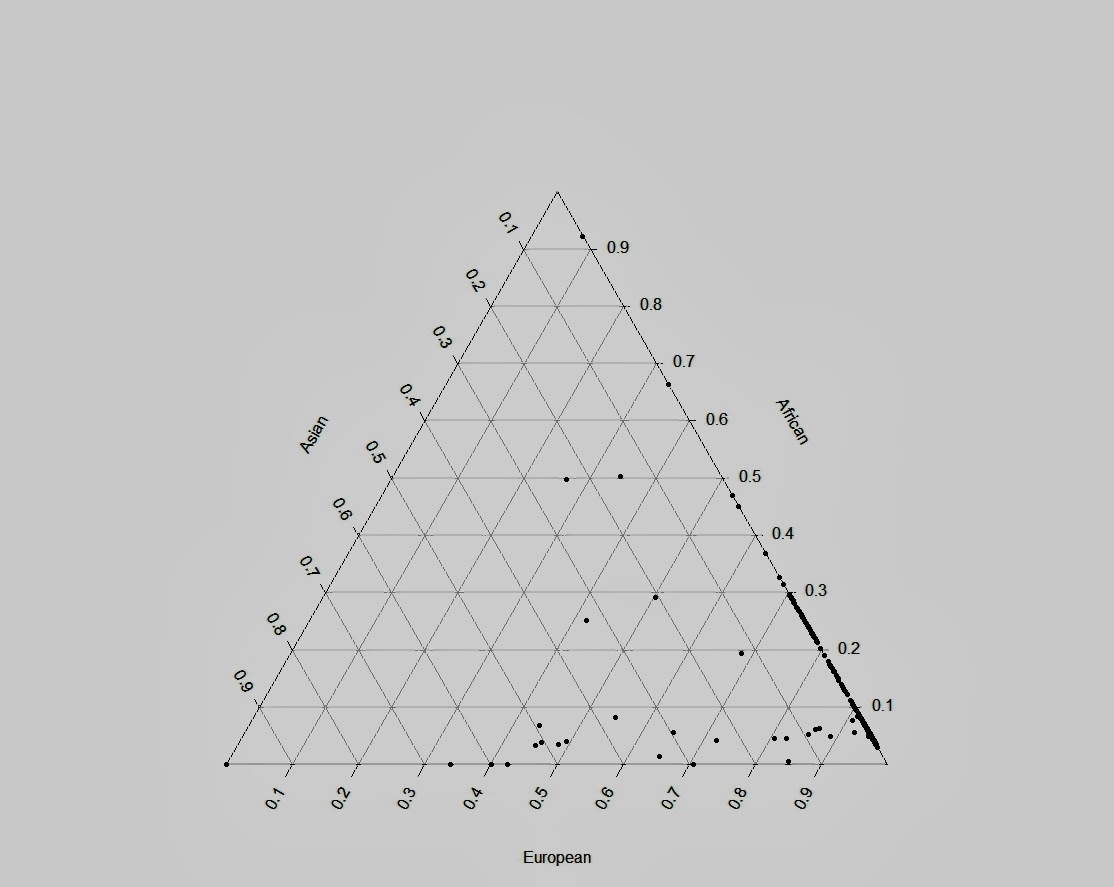
Второй треугольный график, на каждом отрезке которого , представлен процентный вклад одной из трех исконных групп популяции (например: Европы, Африки и Азии, в случае с нашими данными этот пример можно заменить на европейских охотников-собирателей, земледельцев неолита и степных скотоводов эпохи бронзы).
Вот простой код генерирования этих графиков в R. В программе R нет базовых пакетов для построения триангулярных графиков, поэтому нужно будет сначала установить пакет plotrix. Ancestry.txt — это файл полученный на выходе из SNPWEIGHTS:
# EV Plot with Percent Ancestry Overlay
data=read.table("ancestry.txt", as.is=T, header=F)
names(data)
plot(data$EV1, data$EV2, pch=20, col="gray", xlab="EV1", ylab="EV2")
text(data$EV1, data$EV2,labels=round(data$EUR,2)100, cex=0.4, offset=0.1, pos=3)
text(data$EV1, data$EV2,labels=round(data$AFR,2)100, cex=0.4, offset=0.1, pos=2)
text(data$EV1, data$EV2,labels=round(data$ASN,2)*100, cex=0.4, offset=0.1, pos=1)
#Triangle Plot
data$total=data$EUR+data$AFR+data$ASN # Need to account
data$European=data$EUR/data$total # for slight rounding
data$African=data$AFR/data$total # in the ancestry
data$Asian=data$ASN/data$total # estimation file for
data_p=data[c("European","Asian","African")] # triax.plot to work
library(plotrix)
triax.plot(data_p, pch=20, cc.axes=T, show.grid=T)
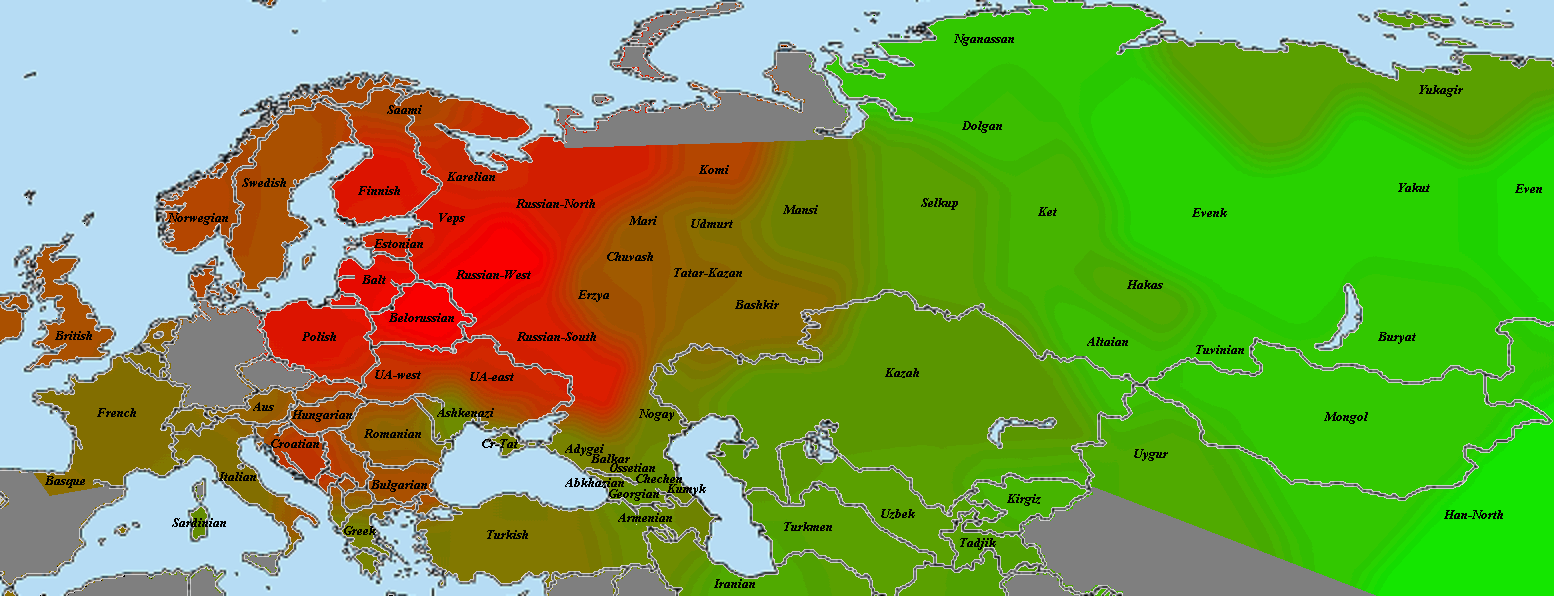
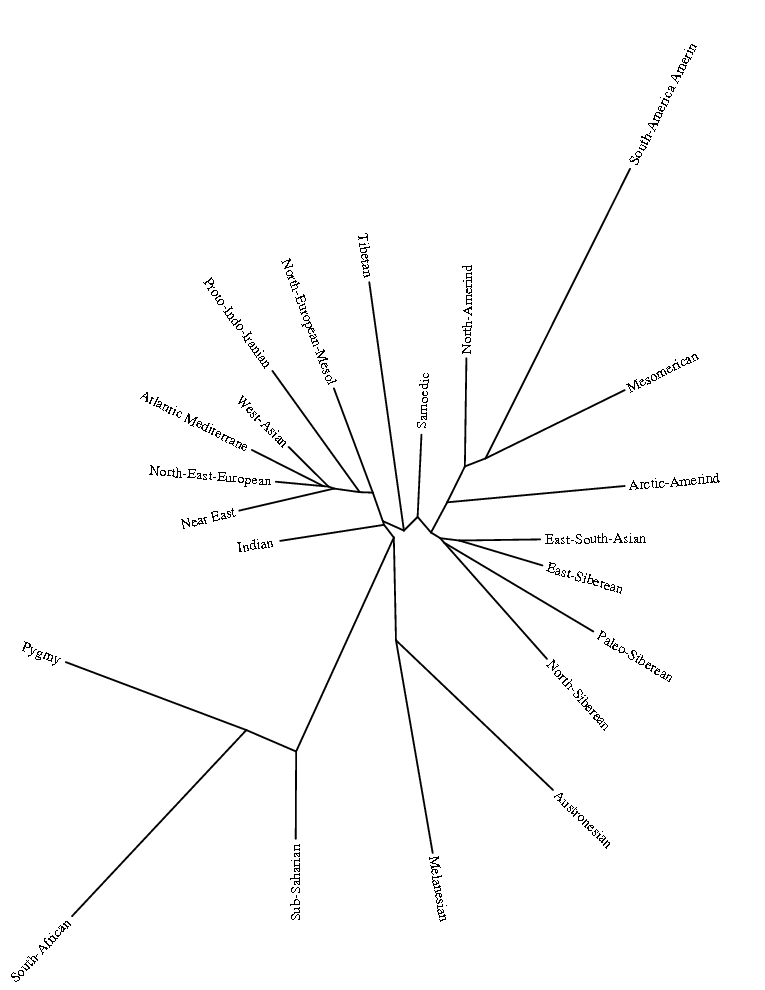
Разумеется, размещенные на сайте разработчика референтные панели носят ограниченный характер. Поэтому я решил заполнить пробелы, преобразовав аллельные частоты SNP-ов в 16 предковых компонентах в 16 синтетических «чистых» предковых популяций, каждая из которых состояла из 200 синтетических индивидов («симулянтов») состоящих на 100 процентов из одного компонента происхождения в модели K16). Файл с генотипами 3200 «симулянтов» я использовал для вычисления весов снипов в каждом компоненте. Продвинутые пользователи, желающие протестировать модель K16 до ее публичного релизма, могут скачать полученный файл с весами снипов здесь, а затем, cледуя приведенным выше инструкциям, использовать его в качестве референтной панели (а затем сравнить свои результаты с усредненными результатами разных этнических популяций).
Я протестировал веса снипов в модели K16 (выражаю признательность автору программу Чену за помощь), и обнаружил, что между данными калькулятора и данными SNPWEIGHTS расхождения носят незначительный характер, хотя похоже, что SNPWEIGHTS не так сглаживает минорные компоненты происхождения (что позволяет легче выделить в пространстве главных компонент кластеры):
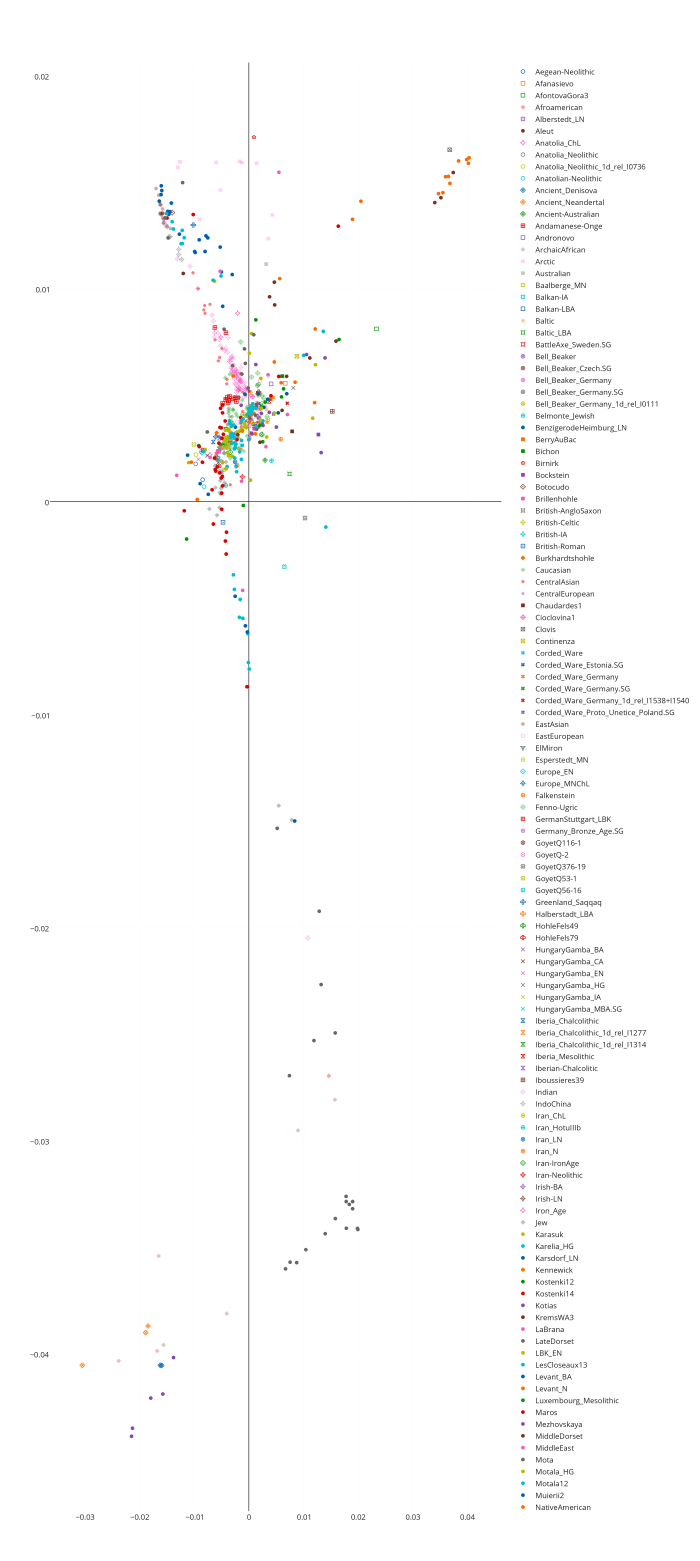




















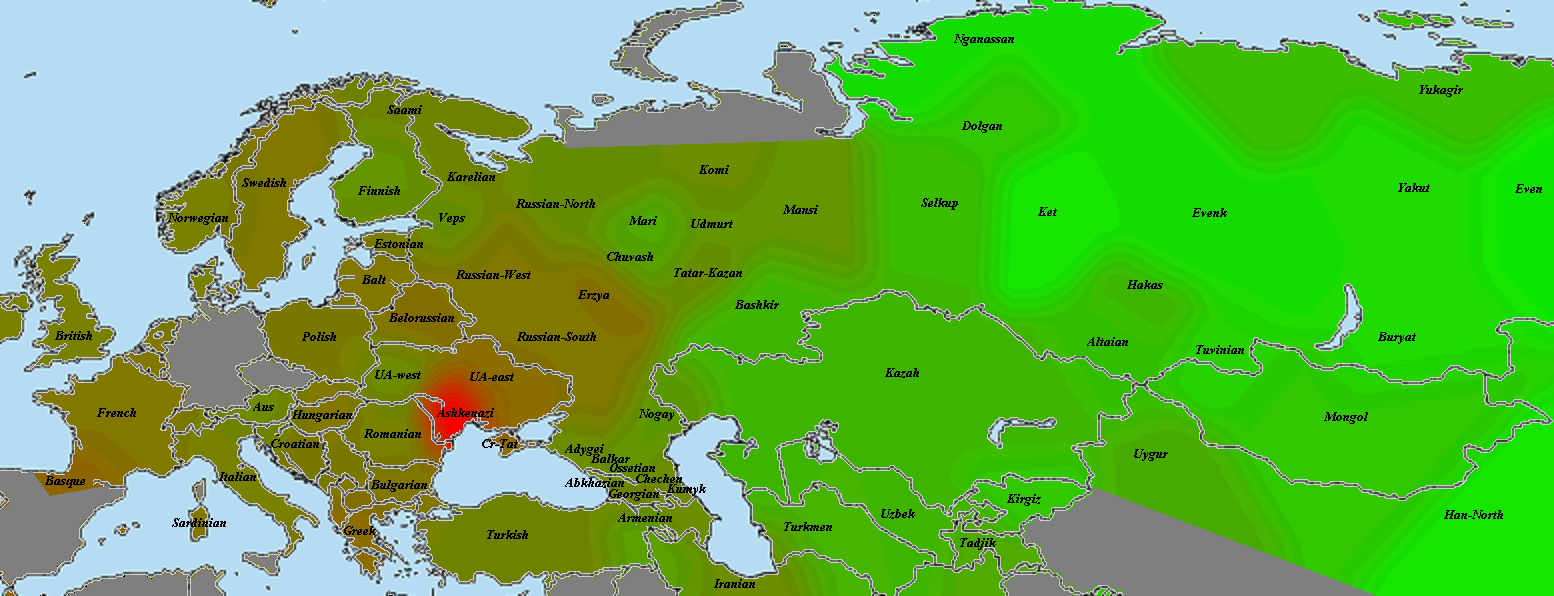
























Для отправки комментария необходимо войти на сайт.