К середине мая этого года я закончил трудоемкий процесс импутации сборной солянки из 9000 публично доступных образцовых представителей 700 различных человеческих популяций, генотипированных в разное время на разных снип-платформах (главным образом — Illumina и Affymetrix)
Строго говоря, я планировал завершить этот этап работы намного раньше, но в ходе выполнения работ возник ряд обстоятельств, помешавших завершить этот этап в срок. Главным из них является смена сервера где я выполнял импутирование геномов. Я начал работать на сервере Мичиганского университета, однако в ходе процесса перешел на аналогичный сервис Института Сэнгера (имени того самого нобелевского лауреата, предложившего первый метод полного сиквенирования генома).
Это решение было продиктовано необходимостью использовать новейшую референсную панель аутосомных гаплотипов — Haplotype Reference Consortium (в нее входит примерно 30 тысяч, а после предстоящего этим летом обновления — свыше 50 тысяч — аутосомных геномов, т.е свыше 60 тыс. гаплотипов). Надо сказать, этнический состав выборки референсных геномов впечатляет, хотя и там по-прежнему наблюдается перекос в сторону европейских популяций. К сожалению, и эта новейшая выборка представлена преимущественно европейцами (поэтому вероятность импутированных генотипов для европейских популяций оказались лучше аналогичных результатов в африканской и азиатской когортах), однако даже с учетом этого обстоятельства ее надежность в определении негенотипированных аллелей снипов выше 1000 Genomes (не говоря уже о HapMap):
| 1 | UK10K | 3715 | 3781 | 6.5x |
| 2 | Sardinia | 3445 | 3514 | 4x |
| 3 | IBD | 4478 | 4478 | 4x + 2x |
| 4 | GoT2D | 2710 | 2974 | 4x/Exome |
| 5 | BRIDGES | 2487 | 4000 | 6-8x (12x) |
| 6 | 1000 Genomes | 2495 | 2535 | 4x/Exome |
| 7 | GoNL | 748 | 748 | 12x |
| 8 | AMD | 3305 | 3305 | 4x |
| 9 | HUNT | 1023 | 1254 | 4x |
| 10 | SiSu + Kuusamo | 1918 | 1918 | 4x |
| 11 | INGI-FVG | 250 | 250 | 4-10x |
| 12 | INGI-Val Borbera | 225 | 225 | 6x |
| 13 | MCTFR | 1325 | 1339 | 10x |
| 14 | HELIC | 247 | 2000 | 4x (1x) |
| 15 | ORCADES | 398 | 399 | 4x |
| 16 | inCHIANTI | 676 | 680 | 7x |
| 17 | GECCO | 1131 | 3000 | 4-6x |
| 18 | GPC | 697 | 768 | 30x |
| 19 | Project MinE — NL | 935 | 1250 | 45x |
| 20 | NEPTUNE | 403 | 403 | 4x |
| Totals | 32611 | 38821 | ||
| 22 | French-Canadian | 2000 | 5-6X | End 2014 |
| 23 | Converge | 12000 | 1x | Now |
| 24 | UG2G Uganda | 2000 | 4x | 2015 |
| 25 | Arab Genomes | 100 | 30x | |
| 26 | Ashkenazi | 128 | CG | Now |
| 27 | INGI-Carlantino | 94 | 4x | Now |
| 28 | CPROBE | 80 | 80 | 4x |
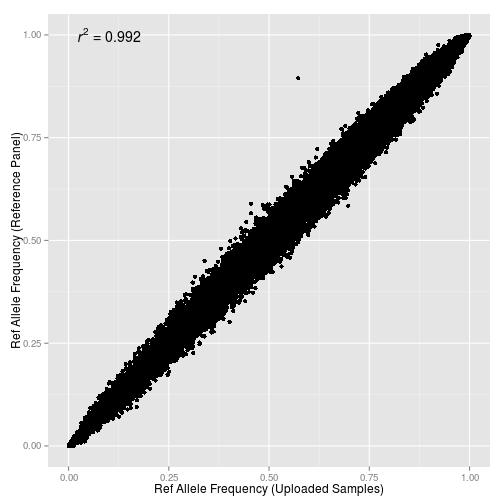
Cледуя рекомендациям, я получил набор из 9000 образцов, каждый из которых включает в себя набор из 20-30 миллионов снипов. К сожалению, из-за субоптимальности результатов в некоторых выборках (Xing et al, Henn et al. и ряде других), их придется исключить из тех видов анализа, которые требует максимальной точности исходных данных. Импутированные генотипы (выраженные через оцененные вероятности) были трансформированы с помощью программы Plink 1.9 в генотипы, причем выбирались варианты полиморфизмов с вероятностью 0.8 (—hard—call—threshold 0.8)
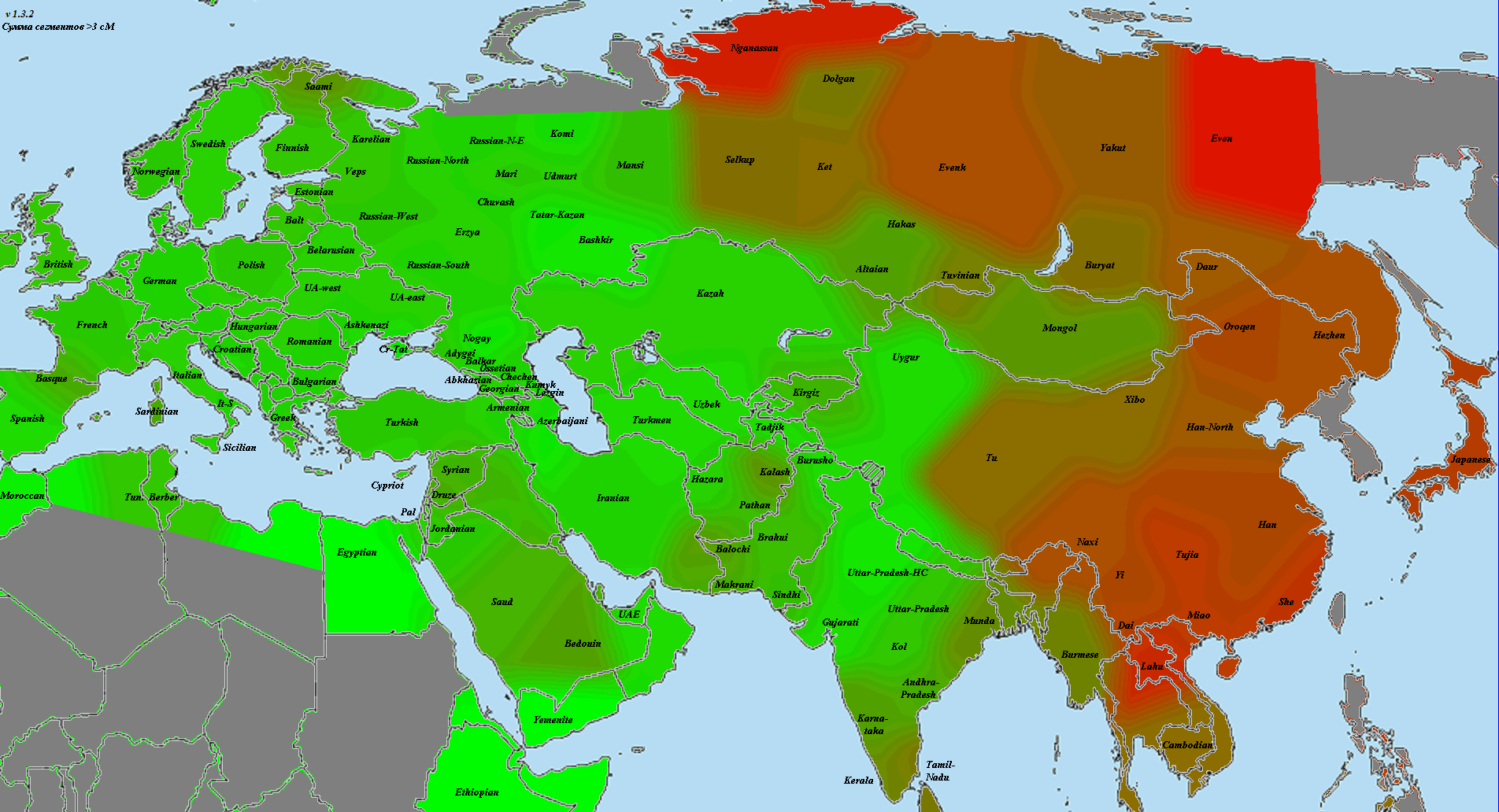
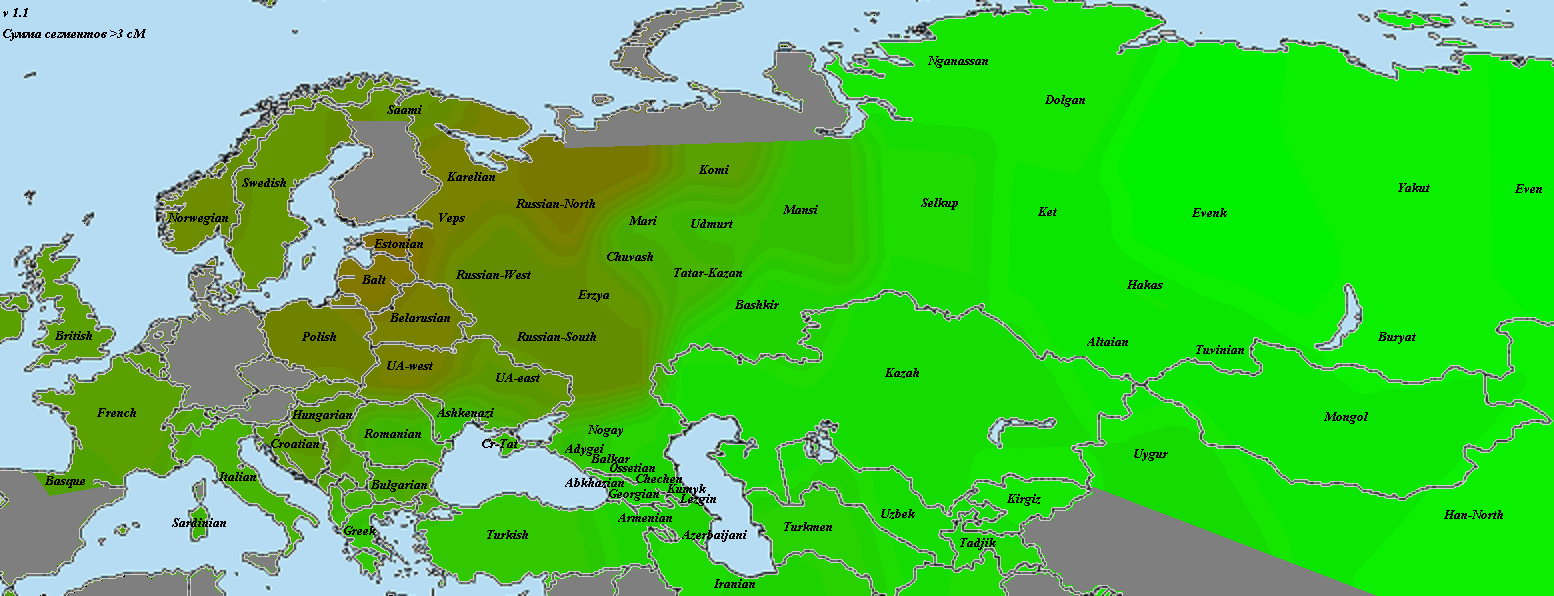
Для оценки полезности импутированных генотипов для популяционного анализа я использовал метрику nearest в программе Plink (матрица с дистанцией между ближайшими геномами) и кластерограммы IBS (идентичности по генотипам).
Таблица метрики nearest (Z-статистика)
А это кластерограмма с хорошо видно географической локализацией кластеров. Я использовал для кластеризации матрицы IBS несколько разных алгоритмов — наиболее убедительный вариант был получен с помощью алгоритма Ward
Другие варианты топологии кластерограмм в формате NEWICK и TRE можно посмотреть здесь (их можно открыть в любой программе для визуализации филогенетических деревьев).
Таким образом, для некоторых типов анализа в популяционной генетике использование импутированных снипов может сослужить хорошую службу, смягчая (или, наоборот, увеличивая) градиент частот аллелей).
Дополнительные анализы — fastIBD, IBS, анализ главных компонентов — образцов в выборке, только подтверждает это наблюдение:

















































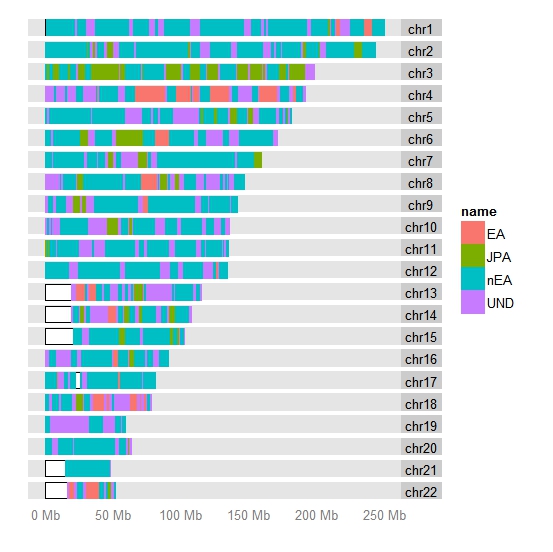
Но самое лучшее подтверждение надежности импутированных снипов для анализа компонентов происхождения было получено с помощью p-теста Z-статистики во время оценки правильности определенной топологии дерева компонентов (с допущением фактора смешивания предковых компонентов). Для этой цели я использовал стандартный инструмент — программу TreeMix. Я использовал только те снипы, которые встречаются в моей контрольной выборке (референсов каждого из компонента) с частотой выше 99 процентов. Как видно из нижеприведенного графика, компоненты выбраны правильно, а топология определяется практически безошибочно, несмотря даже на малое количество снипов (6 тысяч). Правильно определились и направления потоков генов, дрейфов генов (указаны стрелками). Тут в принципе мало нового — большинство этих эпизодов уже были описаны в отдельных работах генетиков. Так, виден поток генов от «денисовского» человека к усть-ишимцу, от которого в свою очередь идет поток генов к австралоидным популяциями. То есть денисовская примесь у папуасов могла достаться от сибирских популяциях близких к «усть-ишимцу». Виден также вклад ANE/EHG в геном североамериканцев -в интервале 10-15 процентов.
Принципально новым является лишь определенный программой дрейф генов в направлении от африканцев Khoisan к североафриканцами (в качестве референса которых взяты египтяне, бедуины и алжирцы). Скорее всего, это и есть тот самый пресловутый сигнал «египтского выхода» человечества из Африки, о котором недавно писалось в новейшей статье, а сам компонент -идентичен пресловутому Basal-Eurasian component
В начале июля в связи с публикацией препринта о генофонде древних ближневосточных земледельцев решился все таки подписать заявление на имя Давида Рейха и Иосифа Лазаридис с ходатайством о доступе к полной версии их выборки (она включает много новых интересных для меня популяций — например, около сотни новых образцов шотландцев, шетландцев, ирландцев из разных областей Ирландии, немцев, сорбов и поляков из восточной и западной Польши).
Г-н Лазаридис был весьма любезен и буквально на следующий день после получения подписанного заявления предоставил мне доступ к этим данным. Я займусь их плотным изучением чуть позже. А пока любопытно посмотреть результаты пилотного Admixture анализа 5900 публичных доступных образцов. В качестве проверки надежности своего нового метода изучения древних и современных популяций людей, я провел 4 параллельных анализа Admixture c разным дефолтным значением предковых популяций (K).
Разумеется, в нашем случае число компонентов K заведомо больше 3, авторы статьи эмпирически показали что меньший разброс значений был получен при K=11. Поэтому я исходил из этой цифры, назначив три разных значения K — 10,11,13.
В первом варианте я использовал т.н unsupervised режим Admixture, т.е. программа должна была сама угадать и реконструировать частоты аллелей снипов в 10 реконструируемых предковых «компонентах» популяций.
Как и ожидалась, таковыми оказались африканский (пик у пигмеев и бушменов), америндский (пик у эксимосов и американских индейцев), сибирский (пиковые значение у нганасанов), южно-индийский компонент (пик в народностях Paniya и Mala), австрало-меланизийский, южно-восточноазиатский, три западно-евразийских компонента — 2 компонента западноевроп ейских и кавказских охотников-собирателей и неолитический; и наконец ближневосточный.
Разумеется, за исключением трех компонентов с пиками в древних геномах, данное распределение отражает cовременное распределение предковых компонентов.
Пришлось вручную выделять из ближневосточного компонента популяцию базальных европейцев (в качестве основы я взял геномы натуфийцев, т.е ближневосточный компонент — Levant_N — может быть разложен на два отдельных предковых компонента — неолитический и мезолитический «натуфийский»), а затем сгенерировать гипотетическую популяцию из 20 образцов состоящих на 100 процентов из натуфийского компонента. Именно этот компонент был включен в модель K11 под названием Levant_Mesolithic ( или Natufian). Этот компонент не стоит путать с компонентом Basal-Eurasian в калькуляторе Eurogenes K7 Basal-rich, так в в моей модели K11 основная часть базального компонента ушла в неолитические компоненты (т.е Natufian=Basal-Rich — Neolithic)
Гораздо сложнее ситуация обстояла с разделением компонента кавказских охотников-собирателей, которые наряду с американскими аборигенами несут в своем геноме значительные доли компонента древних северо-евразийцев. По этому причине очень сложно, например, разделить восточных охотников-собирателей (из мезолитических культур Карелии и Самары) и синхронным им кавказских охотников-собирателей.
Из-за присутствия компонента древних северо-евразийцев в их геноме, в Admixture компонент древних кавказцев увеличивается только за счет компонент восточных охотников-собирателей — и наоборот. Правда, можно попытаться выделить отдельный мезолитический компонент населения горного Загроса (Иран).
В случае успеха древние геномы жителей мезолитической Грузии можно будет представить как 20% компонента степных охотников-собирателей + 80% местного мезолитического субстрата.











































{kind=link}
Для отправки комментария необходимо войти на сайт.