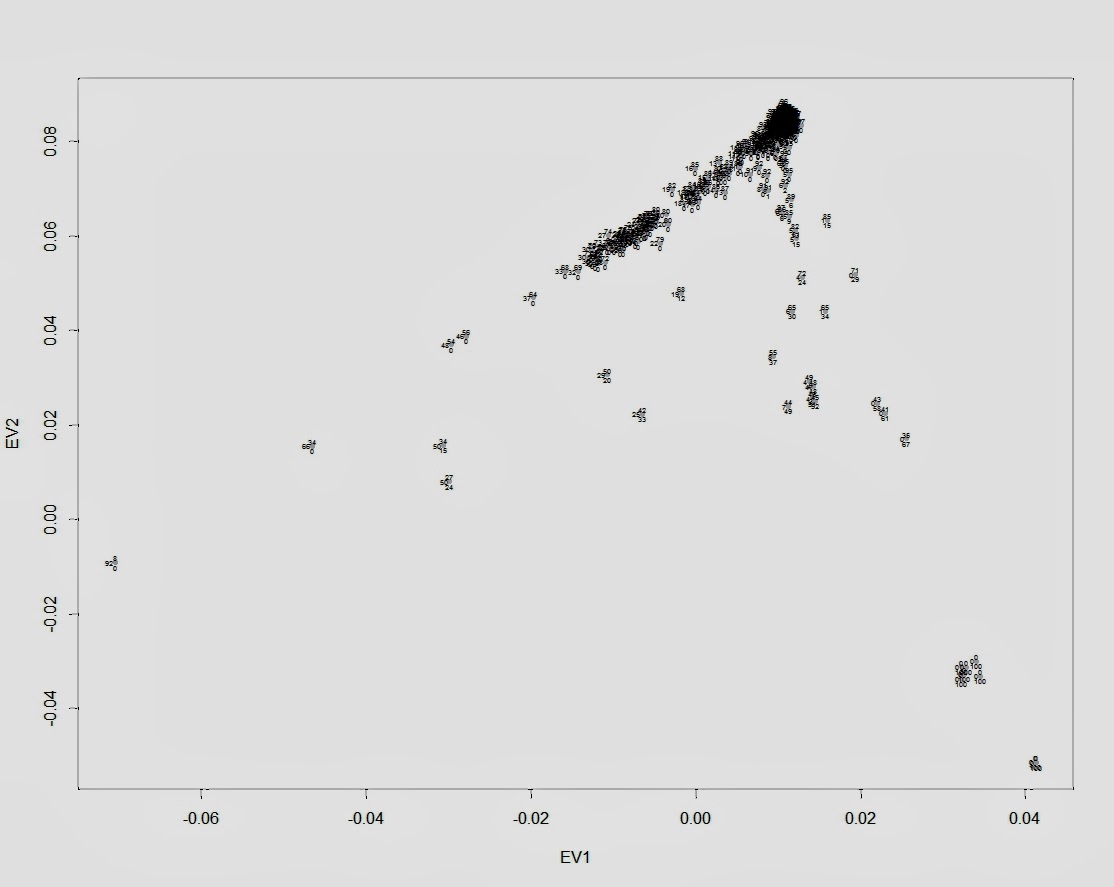
Визуализация количества общих (IBD) сегментов у жителей Европы и Северной Азии.
Этнокалькуляторы на базе Admixture, представляющие результат «просчета» генома испытуемого в виде смеси предковых компонентов, достигли уже очень хорошей точности. Однако у них есть и определенные недостатки. Во-первых, случается, что у двух разных народов пропорции смешения этих компонентов довольно близки, хотя близкого родства между ними не наблюдается. Обычно для исключения такого эффекта увеличивают число компонентов, то есть повышают детализацию. Однако при этом зачастую возрастает и «шумность», случайные отклонения от ожидаемых значений. Кроме того, бывает тяжело понять — смешение произошло в предыдущем поколении, или тысячу лет назад? Если человек происходит из двух отдаленных народов, он часто позиционируется в географической точке, находящейся между ними, и непохож ни на один из родительских народов. При более сложносоставном происхождении все запутывается еще сильнее.
Нет ли метода напрямую измерить уровень родства отдельного человека с той или иной популяцией? При такой постановке вопроса сразу приходит на ум один из возможных ответов — необходимо просчитать количество IBD (то есть идентичных благодаря общности происхождения) аутосомных сегментов. Такой подход уже реализован в утилите от 23andMe под названием Countries of Ancestry, однако с рядом заметных недочетов. Используются результаты опроса пользователей сервиса о стране их происхождения, при этом непонятно, каков размер выборки от каждой страны. Да и детализация уровня «страна» для жителей России явно не подходит — зачем мешать в одну кучу карел, осетинов и якутов.
К счастью, эти проблемы можно частично устранить, используя научные выборки (либо коммерческие, однако набрать подобный объем из коммерческих выборок мне сейчас не по силам). С удешевлением процесса генотипирования количество имеющихся в открытом доступе выборок начало быстро расти. В первую очередь я использовал выборки, выложенные на сервере Эстонского биоцентра . Они стали основой. Часть пробелов была заполнена выборками из недавней работы Hellenthal , их пришлось переконвертировать из build 36 в build 37. Отдельное спасибо Вадиму Вереничу за помощь с несколькими выборками, хорошо увеличившими охват этнокарты.
Главной сложностью в работе оказалось сведение геномов из всех источников вместе. В каждой научной работе использовался свой набор снипов, часто с разной ориентацией. Коммерческие выборки тоже неоднородны — например, в FTDNA, как оказалось, существует четыре варианта файлов raw data со слегка отличающимся набором снипов и разной ориентацией примерно трех сотен из них. Добавьте к этому два варианта выравнивания и трансферы из 23andMe (у которой нашлись свои заморочки, например, дублирование одних и тех же снипов под разными названиями).
Конечно, хотелось использовать как можно большее количество снипов. Однако после ряда попыток придумать коэффициенты пересчета и прочее, стало понятно, что это методологически неверно. Пришлось оставить лишь те снипы, которые присутствовали во всех используемых выборках, в стандарте FTDNA, а также на чипе v3 от 23andMe. Вероятно, в будущем придется включить в просчет и новый, четвертый чип от этой компании, однако пока я решил с ним не связываться. В общем итоге осталось около 244 тысяч снипов — не так уж мало, я опасался худшего. От покрытия FTDNA это составляет чуть больше трети.
Компания FTDNA и сервис Gedmatch используют для фильтрации общих сегментов критерий наличия не менее 700 снипов. Однако для мелких сегментов он выполняется не так уж часто (из-за чего у клиентов FTDNA возникает иллюзия сравнительно небольшого количества таких сегментов). Поэкспериментировав, я остановился на рубеже в 150 снипов — менее него количество сегментов, являющихся статистическими артефактами, начало быстро расти. Основным показателем для отрисовки на этнокарте я взял общую сумму сегментов длиной более 3 сМ. Конечно, более длинные сегменты являются более четким показателем родства, однако их заметно меньше. А это значит, что их количество более подвержено случайным отклонениям. С другой стороны, более мелкие сегменты сливаются в общую кашу. Таким образом, выбранный критерий является компромиссом. При увеличении объема выборок на порядки станет возможно использовать только длинные сегменты и улавливать родство более четко.
Метод дает релевантные результаты при сравнении с выборками свыше 10 человек. Чем меньше размер выборки, тем сильнее влияние случайных отклонений. Из-за этого часть выборок я объединил вместе (например, литовцы и латыши стали балтами), часть исключил с карты. Однако некоторые все же пришлось оставить — в первую очередь это финны (2 человека), западные украинцы (6), башкиры (6) и австрийцы (4). Если для какой-то популяции значения явно выпадают из ряда соседей, всегда обращайте внимание на размер выборки, приведенный в сопроводительной таблице.
Одновременно достоинством и недостатком метода является сильное влияние «эффекта основателя», «множественного родства», «бутылочных горлышек» и т.д. За этим перечислением скрывается примерно одно и то же — когда популяция происходит от сравнительно небольшой группы людей, ее члены разделяют между собой большое количество общих сегментов. Наиболее известным примером являются евреи-ашкенази — достаточно иметь одного отдаленного предка из этого народа, чтобы получить множество генетических «кузенов». Таким образом, родство с народом, подвергшемуся такому эффекту, видно более четко. Но это же искажает общую картину — одинаковое количество генетических пересечений может означать совершенно разную степень близости в зависимости от истории популяции.
Я сравнил 26 человек из коммерческих выборок, представляющие различные популяции интересующих меня регионов, с набором из 1130 геномов, взятых из научных выборок. Результаты сведены в таблицу и частично визуализированы на картах. При интерпретации помните о вышеперечисленных искажениях!
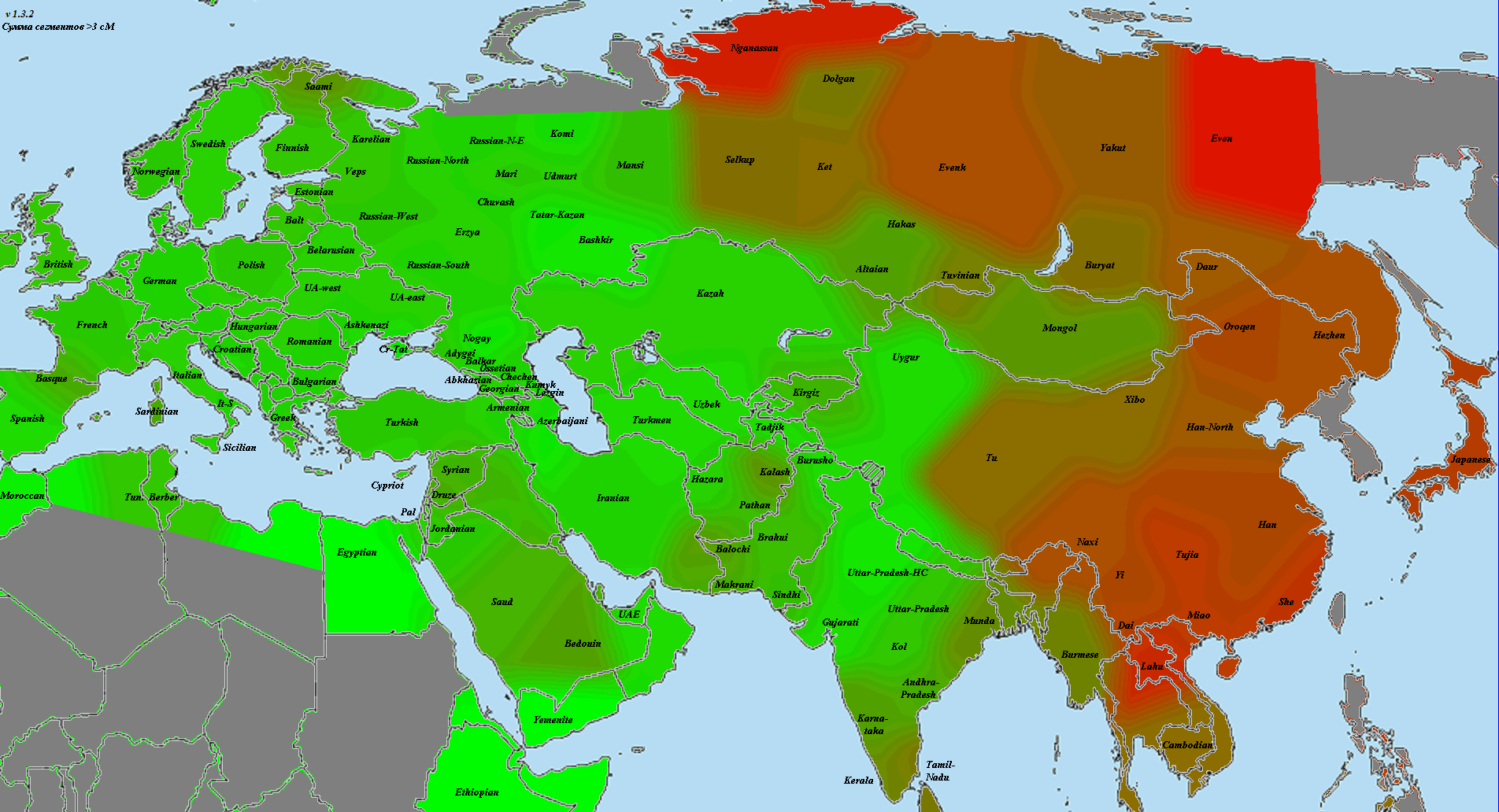
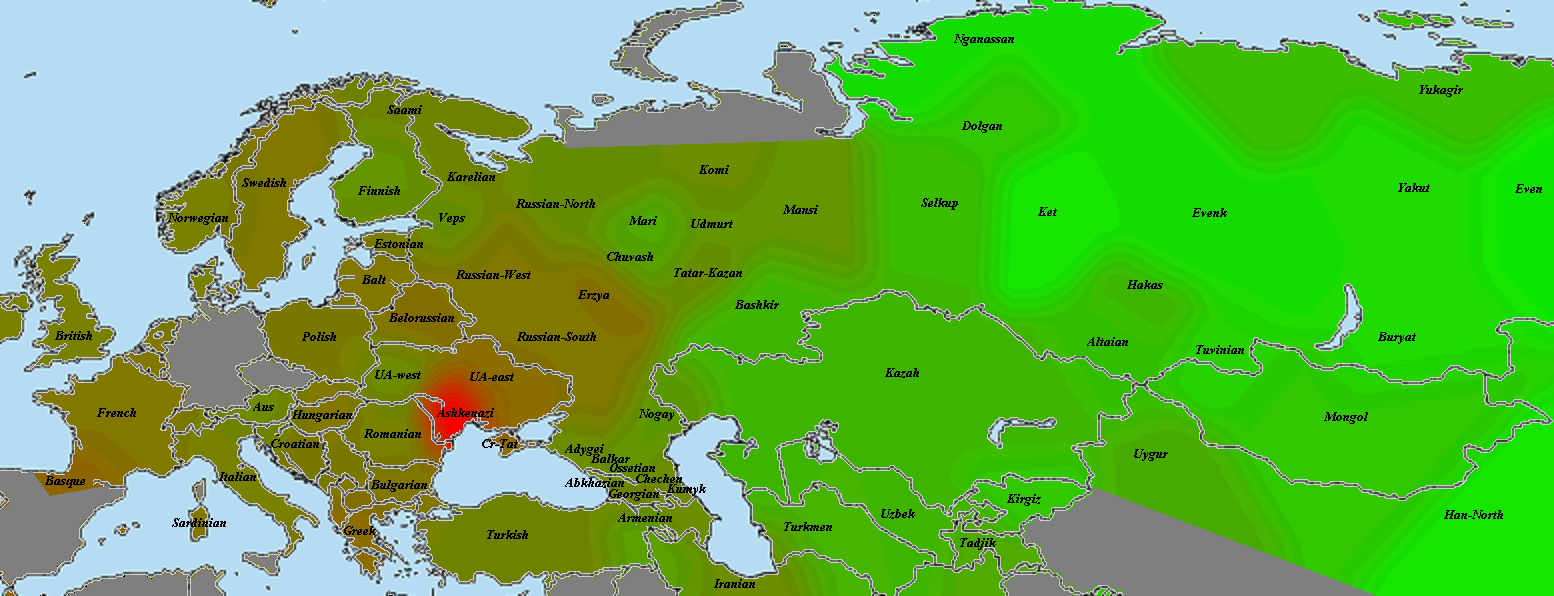
Начнем с представителя народа, считающегося наиболее архетипичными восточноевропейцами в большинстве этнокалькуляторов. Это литовцы (картинка увеличивается по клику):
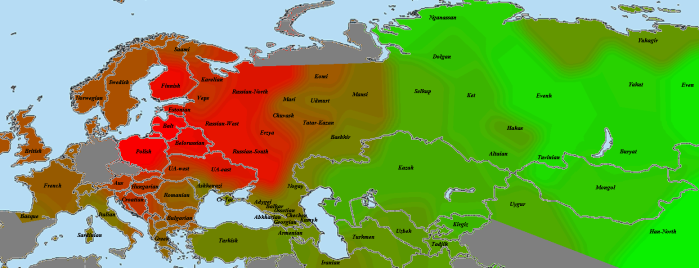
Как видно, литовец оправдывает это звание и по количеству общих сегментов. Красное пятно закрывает большую часть Восточной Европы, в том числе и балтийских финнов. Условно говоря, на этой карте мы видим некий «базовый уровень родства» среди восточноевропейцев.
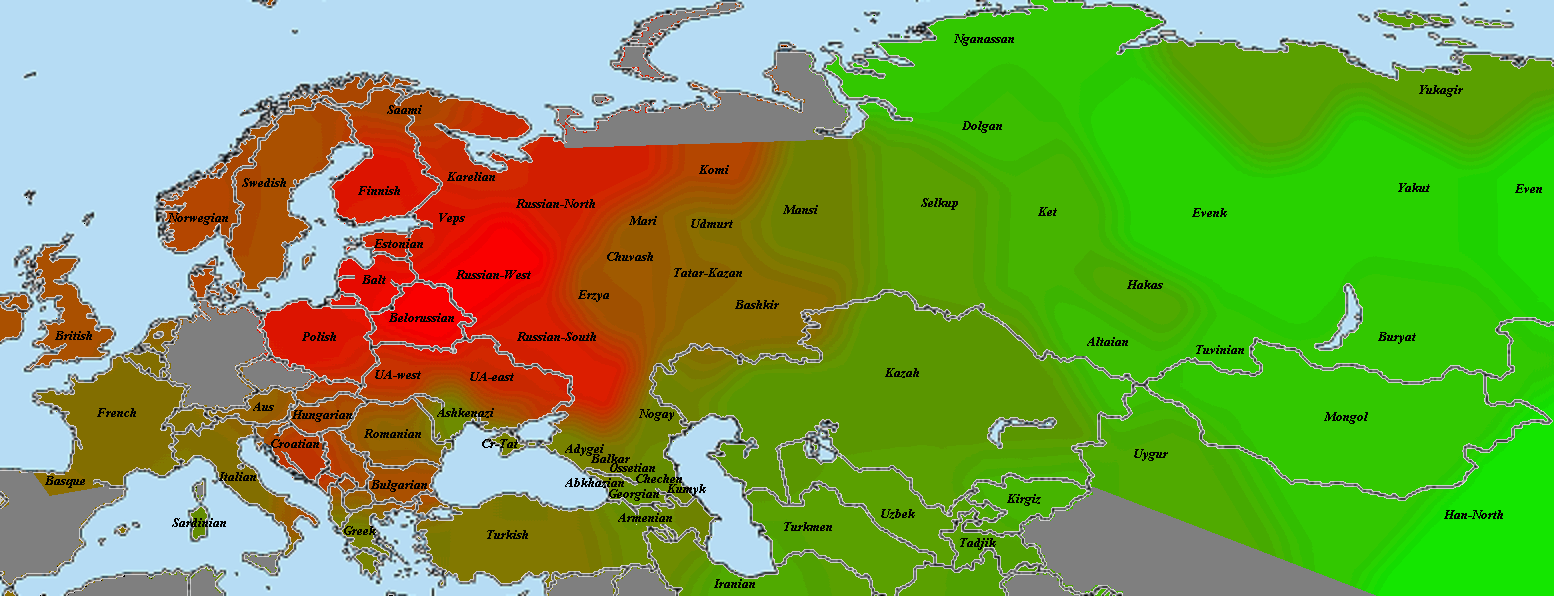
Пятно восточного финна практически совпадает по форме, однако распределение интенсивности иное:

Я бы сказал, что в основном это более частный и специфичный вариант того же, что мы видим у литовца. Доказательством может служить высокий уровень пересечения с балтской выборкой. В то же время, существует и финская специфика, например, пересечение с саами, которые у литовца довольно бледные. Более яркое и пересечение со шведами. Скорее всего, здесь мы видим результат включения в состав шведов финского субстрата, поскольку с теми же норвежцами интенсивность явно ниже.
Крайней западной точкой у нас будет представитель российских немцев. На этнокалькуляторах Admixture он получается достаточно типичным представителем немецкого народа, поэтому версию о заметном влиянии на его наследственность русских можно исключить.
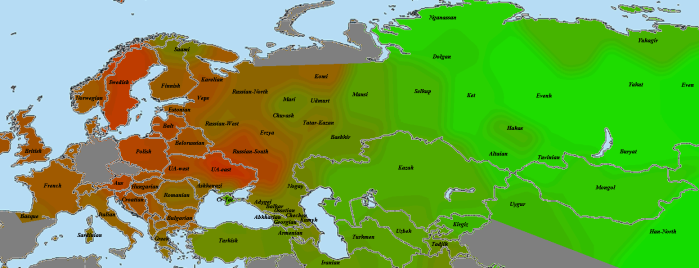
К сожалению, немецкой выборки у меня нет, поэтому Германия закрашена серым. Некоторым заменителем является Швеция, которая чуть ярче соседей. К некоторому удивлению, французы и британцы не показали заметной общности с немцем, хотя ее уровень все же выше средневосточноевропейского. Частично это может объясняться тем, что в британской выборке лишь семь человек из 23 — англичане, остальные являются ирландцами. шотландцами и валлийцами. Пятно у восточных украинцев и южных русских также загадочно — неужели это след знаменитых готов?
Невозможно исследовать генетическое разнообразие восточноевропейцев и обойти при этом ашкенази. Поэтому я позволил себе небольшую некорректность и разместил их на карте в районе нынешней Одессы. Картинка для ашкенази из коммерческой выборки:
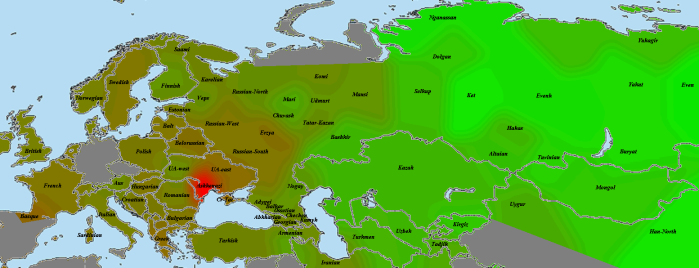
Ожидаемое ярко-красное пятно сходства с родной популяцией, остальные все довольно далеко (на втором месте получилась выборка сефардов, но ее на карте нет). Повышение у басков и греков показывает родство ашкенази со средиземноморскими популяциями, пятно у восточных украинцев и белорусов объяснимо длительным совместным проживанием.
Перейдем к восточным славянам. Небольшой размер выборки западных украинцев не помешал им оказаться на первом месте у карпатского русина:

Пятна на остальной территории получились довольно неровными. Я бы не стал делать из этого каких-то глубоких выводов о древних пересечениях карпатцев и финнов или эрзян.
Северо-восточная Беларусь:

Украина (Полтава):
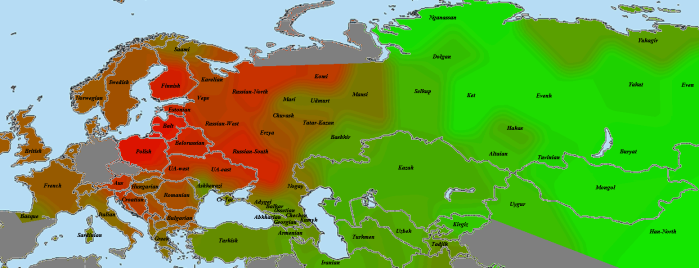
Обращает на себя внимание пересечение с поляками.
Человек смешанного происхождения — донские казаки и украинцы:
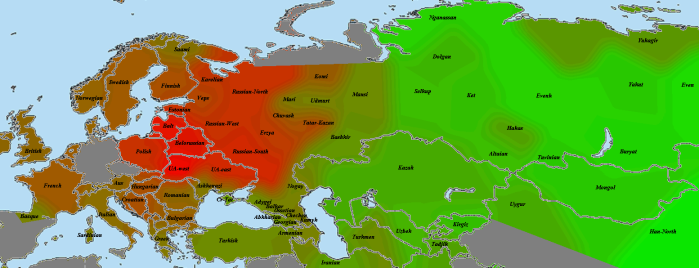
Тверь-Рязань:

Как видно, балто-славянская общность улавливается всегда, в то время как более тонкие различия частично видны, частично скрываются шумом (случайными отклонениями).
Представители эрзи и мокши явно в своей основе близки балто-славянам. При этом балтийские финны никак не выделяются, а народы волго-уральского региона уже довольно далеки. Все это не является новостью для интересующихся темой людей, однако независимое подтверждение результатов показывает действенность методики.
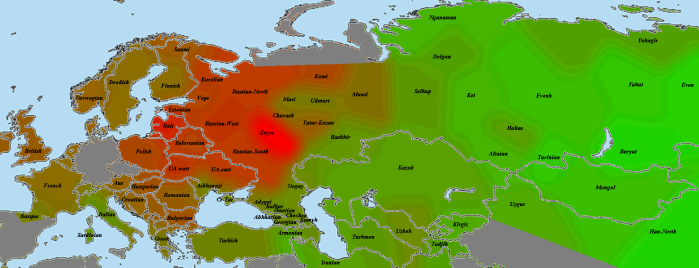
Эрзя:
Мокша:
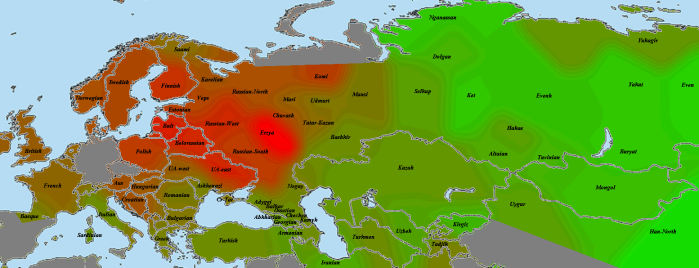
Мокшанская выборка не помещена на карту из-за своего маленького размера (давала слишком большие случайные отклонения). У мокши «родная» выборка получилась заметно ближе эрзянской, у эрзи, соответственно, наоборот. Вероятно, это значит, что, несмотря на родственность двух групп, различие между ними с точки зрения разделяемой популяционной истории существенно (простыми словами, женились преимущественно внутри своего народа).
У северного русского видно родство как с балто-славянскими выборками, так и с балтийскими финнами:

Наряду с этим, у русского из Пермского края ощущается влияние коми. Вероятно, с этим же связано и приближение других народов Урала:

В то же время, сами коми-зыряне скорее относятся к тому же «балто-славяно-финскому» кругу популяций:

Чтобы не делать пост бесконечным, карты для народов волжско-уральского региона (в широком смысле) будут приведены в следующей части.












































































Для отправки комментария необходимо войти на сайт.