
Закончил на 99% подготовку 2 моделей этно-популяционных калькуляторов ДНК — заточенную под deep ancestry (анализ современных геномов с использование древних геномов) K11 и модель для анализа популяционного происхождения современных популяций K16.
В число 16 «предполагаемых предковых» популяций в K16 входят следующие выделенные группы:
Австрало-веддоидная
Палеолитические охотники-собиратели Кавказа
Американские аборигены
Охотники-собиратели скандинавского мезолита
Австронезийцы
Ближневосточные неолитические земледельцы
Сибирские аборигены
Ближне-восточные популяции
Североафриканские популяции
Популяции западной Африки
Северные популяции Индостана
Юго-восточноазиатские популяции
Восточные охотники-собиратели
Неолитическое население Европы
Восточно-африканские популяции
Западноевропейские охотники-собиратели
Таблица FST между компонентами K11 (FST — Индекс фиксации Райта Fst, отражающий меру дифференциации популяций)
Кластеризация компонентов модели K11 по степени дифференциации
Таблица FST между компонентами K16
Кластеризация компонентов модели K16 по степени дифференциации
На следующем PCA графике отображены 2 группы компонентов — предковые компоненты K16 (полученные в программе ADMIXTURE в ходе анализа современных популяций) и предковые компоненты K11 (они вычислены в той же программе, но на другой выборке аутентичных палеогеномов). Поскольку у пользователей подобных калькуляторов часто возникает вопрос о соотношении компонентов разных моделей калькуляторов, я решил разместить их на одном графике. Методология довольно проста. Сначала я сгенерировал в программе PLINK 220 «синтетических» геномов (20 индивидов в 11 группах). В основу положен предложенный Понтикосом метод популяционных «zombies», в котором используется частоты аллелей снипов, полученных в программе ADMIXTURE. Каждая из 11 групп состоит из 20 «индивидов», геном которых на 100% состоит из одного компонента.
То же самое я сделал с компонентами K16. Затем в целях изучения соотношения компонентов этих двух разных моделей, я пропустил «геномы синтетических индивидов» K16 через калькулятор K11. В итоге выяснилось, что только несколько компонентов K16 полностью совпадают с компонентами K11 (например, Amerindian и African). Остальные компоненты K16 разложились на комбинации компонентов K11. Этот простой эксперимент еще раз подтвердил очевидный факт: предковые компоненты ADMIXTURE, выявленные в ходе анализа современных популяций только в редких случаях соответствуют настоящим предковым компонентам. Большинство подобных компонентов возникают в результате сложного процесса фиксации аллельных частот, например в тех случаях, когда непосредственно после смешивания предковых групп разного происхождения происходит процесс генетического дрейфа. Закон Харди—Вайнберга утверждает, что в теоретической идеальной популяции распределение генов будет оставаться постоянным из поколения в поколение. Так, в популяции растений количество «внуков» с генами высокорослости будет ровно таким же, сколько было родителей с этим геном. Но в реальных популяциях дело обстоит иначе. Из-за случайных событий частота распределения генов из поколения в поколение несколько варьирует — это явление называется дрейфом генов. Рассмотрим крупную размножающуюся популяцию со строго определенным распределением аллелей. Представим, что по той или иной причине часть этой популяции отделяется и начинает формировать собственное сообщество. Распределение генов в субпопуляции может быть нехарактерным для более широкой группы, но с этого момента и впредь в субпопуляции будет наблюдаться именно такое, нехарактерное для нее распределение. Это явление называется эффектом основателя.Дрейф генов сходного типа можно наблюдать и на примере явления с запоминающимся названием эффект бутылочного горлышка. Если по какой-либо причине численность популяции резко уменьшится — под воздействием сил, не связанных с естественным отбором (например, в случае необычной засухи или непродолжительного увеличения численности хищников), быстро появившихся и затем исчезнувших, — то результатом будет случайное устранение большого числа индивидуумов. Как и в случае эффекта основателя, к тому времени, когда популяция вновь будет переживать расцвет, в ней будут гены, характерные для случайно выживших индивидуумов, а вовсе не для исходной популяции.
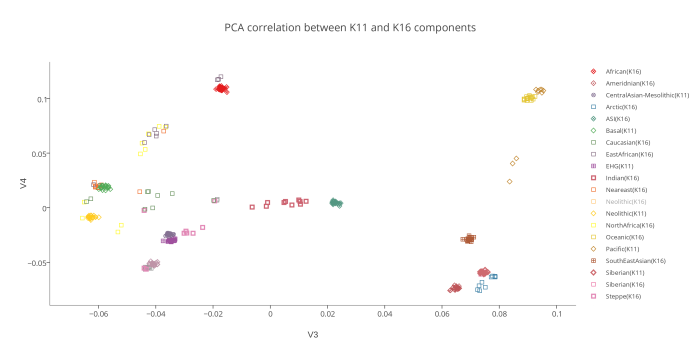 Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).
Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).
Для облегчения понимания сказанного, приведу немного теории. Начну с основ.
Определение базовых терминов
ADMIXTURE (буквально: примесь) – это компьютерная программа (анализ), позволяющая выявлять смешанность состава некоего набора индивидов на основе данных о генотипах и тем самым строить предположения о происхождении популяции.
Принцип работы ADMIXTURE.
Рассмотрим принцип работы ADMIXTURE на примере образцов и популяций из проекта HapMap.
Всего у нас N = 324 образца/индивида, каждый из которых относится к одной из четырех нижеперечисленных популяций:
АФРИКА (ASW) – Африканские предки из Юго-Западной части США
ЮТА (CEU) – жители штата Юта США с корнями из Северной и Западной Европы
МЕКСИКА (MEX) – Мексиканцы, Лонг-Айленд США
ЙОРУБА (URI) – Йоруба, Нигерия
Для удобства дальнейшего изложения будем называть эти популяции «известными».Также мы предполагаем, что они произошли от К разных предковых популяций (мы не знаем от каких именно). В дальнейшем будем называть эти предковые популяцие «предполагаемыми предковыми». Этих «предполагаемых предковых» популяций на самом деле не существует, у них нет общепризнанных названий и характеристик. И на этом этапе мы даже не знаем какие образцы к какой из этих К популяций могут быть отнесены. Теоретически возможно, что образцы из одной и той же «известной» популяции могут принадлежать к двум разным «предполагаемым предковым» популяциям.
Пример 1.
Предположим, что К = 3.
ADMIXTURE далее работает с образцами (их генотипами) и заданным нами числом К = 3. Имея сведения о генотипах и предположение о количестве «предполагаемых предковых» популяций (К) ADMIXTURE строит свою модель (предположение) того, каков вклад каждой из «предполагаемых предковых» популяций в каждый индивид. В результате мы имеем для каждого индивида 3 цифры: количественный вклад каждой из трех популяций (или образно говоря, на сколько процентов данный индивид состоит из первой «предполагаемой предковой» популяции, на сколько – из второй и на сколько – из третьей). При этом может быть и такая ситуация, что у конкретного индивида в составе отсутствует какая-то из «предполагаемых предковых» популяций, даже возможно, что он принадлежит только к одной из «предполагаемых предковых» поуляций. Предположим, для индивида №1 эти цифры такие: 0.3, 0.5 и 0.2. Что эти цифры означают? Означают они доли каждой из «предполагаемых предковых» популяций (ППП) в индивиде №1, т.е. индивид состоит на 30% из первой ППП, на 50% — из второй и 20% — из третьей. Чем больше вклад каждой ППП в индивида, тем больше индивид является «носителем» данной популяции и ее представителем.
Так называемый этно-популяционный калькулятор ДНК представляет собой инструмент, позволяющий использовать заранее определенные (вычисленные) компоненты этнического происхождения K для определения той комбинация исходных предковых компонентов дает наилучшее соответствие (аппроксимирует) происхождение носителя тестируемой ДНК.При создании калькулятора ДНК в основу берется определенная модель (например, задается исходное число компонентов или состав референсной выборки), что неизбежно приводит к определенным уступкам в плане точности и проявлению слабых сторон модели. Например, часто люди критикуют подобные модели калькуляторов за излишнюю европоцентричность и недостаточную представленность геномов из других мест, или же используемые для определения компонентов происхождения выборки данных по отдельным популяциям слишком малы для определения сложной субструктуры генофонда референсной популяции. Наконец, более грамотные люди указывают на отсутствие необходимо инструментария (например, формальной статистики) для проверки статистической значимости определенных компонентов в отдельных моделях калькулятора.
Движок обеих калькуляторов — все та же программа DIYDodecad, После того, как ппрограммма ДНКа калькулятора выдаст первичные результаты — процентное распределение компонентов этно-популяционного происхождения в изучаемом геноме, можно будет перейти к вторичному анализу. Суть его проста — зная процентную комбинацию компонентов происхождения в своем геноме, довольно просто смоделировать свой геном в виде смеси нескольких референсных популяций.
Поэтому, в отличие от предыдущих релизов, K11 и K16 будут включать в себя дополнительный контент:
1) классический Oracle, позволяющий смоделировать анализируемый «геном» (точнее, набор из 100-200 тысяч информативный снипов) в виде комбинации двух референсных популяций, а также установить группу генетически ближайших референсных популяций к геному изучаемого индивида. Однако этот инструмент не может быть использован в случае сложного смешанного происхождения (например, когда изучаемый индивид происходит из более чем двух разных этнических популяций). Иногда программа выдает довольно глупые комбинации, cущественным образом понижая достоверность результатов. Впрочем основное преимущество Oracle и состоит в том, что программа предлагает вместо окончательного «простого» решения список альтернативных вариантов.
Пример: в качестве примера я буду использовать собственные данные.
Исходя из полученных в модели K16 значений компонентов, мой условный наиболее близок к восточнославянским популяциям
«Ukrainian-Center» «2.5884»
«Pole» «3.0962»
«Sorb» «3.1733»
«Polish_West» «3.5992»
«Russian-North-West» «3.7265»
«Russian_Smolensk» «3.834»
«Polish» «4.0348»
«Belarusian_EastBelarus» «4.0852»
«Belarusian_WestBelarus» «4.1216»
«DonKuban_cossack» «4.7769»
В комбинированном варианте двух смешанных популяций распределение предковых компонентов происхождения может быть аппроксимировано следующими комбинациями:
«65.8% Belarusian_EastBelarus + 34.2% Norwegian» «1.1023»
«66.4% Belarusian_EastBelarus + 33.6% Icelandic» «1.1118»
«80.9% Latvian + 19.1% Spanish_Baleares_IBS» «1.1154»
«30% French + 70% Lithuanian» «1.1206»
«29% French + 71% Latvian» «1.1215»
«55% French_West + 45% Lithuanian_Zemajitia» «1.1302»
«28.9% French_East + 71.1% Latvian» «1.1402»
«29% French_Northwest + 71% Latvian» «1.1563»
«72.3% Belarusian_EastBelarus + 27.7% Orcadian» «1.1766»
«57.2% European_Utah + 42.8% Lithuanian_Zemajitia» «1.1825»
Основная часть генома — условно славяно-балтийская (что ожидаемо), но с существенным сдвигом в сторону Скандинавии и западной Европы(примерно 20-30%). Скорее всего, это наследие готов, или контактов балтийских племен с викингами. Интересно, что модель K11 (c использованием современных референсных популяций) дает примерно такой же расклад — разве что древний скандинавско-германский пласт выражен чуть резче чем в модели K16
«Belarusian_West» «2.3841»
«Belarusian» «2.4187»
«Pole_Poland» «2.5278»
«Belarusian_East» «3.7288»
«Russian_Central» «3.7635»
«Swede» «3.9724»
«Russian_cossack» «4.1139»
«Ukrainian» «4.2647»
«Russian_Southern» «4.5204»
«Ukrainian_East» «4.8635»
«66.6% Icelandic + 33.4% Latvian» «1.586»
«41.1% Latvian + 58.9% Orcadian» «1.5898»
«47.9% Lithuanian + 52.1% Orcadian» «1.6007»
«60.2% Icelandic + 39.8% Lithuanian» «1.6082»
«5.7% Basque_Spanish + 94.3% Belarusian» «1.6386»
«5.8% Basque_French + 94.2% Belarusian» «1.6406»
«67.2% Belarusian + 32.8% Swede» «1.659»
«40.2% Lithuanian + 59.8% Norwegian» «1.6876»
«33.7% Latvian + 66.3% Norwegian» «1.689»
«94.1% Belarusian + 5.9% Spanish_Pais_Vasco_IBS» «1.7359
В палеокалькуляторе K11 (т.е. с древними геномами) картинка кажется более убедительной
«Unetice_EBA» «2.7065»
«Bell_Beaker_Czech» «5.0633»
«British_AngloSaxon» «5.1998»
«Nordic_LN» «5.6157»
«Corded_Ware_Proto_Unetice_Poland» «6.3751»
«Nordic_MN_B» «6.3865»
«Halberstadt_LBA» «6.4422»
«BenzigerodeHeimburg_LN» «7.4695»
«Nordic_IA» «7.5404»
«Corded_Ware_Estonia» «7.7635»
Из всех палеогеномов наиболее близок к моему геном представителя унетицкой культуры. Происхождение унетицкой культуры до сих пор не выяснено. Между позднейшими энеолитическими культурами и унетицкой культурой существует типологический и хронологический разрыв. Наибольшее признание в результате последних исследований получило предположение, согласно которому в ее возникновении главную роль сыграли культура колоколовидных кубков и надиревская культура, распространенная в Венгрии (см. ниже). У культуры колоколовидных кубков и унетицкой имеется сходство в керамике, в погребальном обряде и в орудиях труда. Небольшую роль могла сыграть культура шнуровой керамики, хотя в целом они очень различаются. Закономерно, что следующими — хотя и с большим отрывом — близкими к моему геному группами палеогеномов являются геномы древних англосаксов (которые близки к древним скандинавам) и представителей чешского ареала культуры колоковидных кубков).
Аналогично, в режиме смешенных популяций хорошо заметны две тенденции. Во-первых, мой геном может быть представлен в виде комбинации палеогенома представителя позднебронзового века (Хальберштадт) и палеогеномов восточных охотников-собирателей эпохи энеолита, во-вторых как смесь 23.4% генома представителей балтийской позднебронзовой эпохи и все того же позднебронзового палеогенома из Хальберштадта
«86.4% Halberstadt_LBA + 13.6% Karelia_HG» «2.139»
«74.1% Bell_Beaker + 25.9% LesCloseaux13_Mesolithic» «2.1574» «35.9% Hungary_BA + 64.1% Poltavka_MBA_outlier» «2.319»
«65.7% Halberstadt_LBA + 34.3% Poltavka_MBA_outlier» «2.4387»
«83.2% Alberstedt_LN + 16.8% Karelia_HG» «2.443»
«23.4% Baltic_LBA + 76.6% Halberstadt_LBA» «2.4846»
«16.7% Europe_MN + 83.3% Poltavka_MBA_outlier» «2.4897»
«83.4% Halberstadt_LBA + 16.6% Samara_Eneolithic» «2.536»
«12.9% Halberstadt_LBA + 87.1% Unetice_EBA» «2.5603»
«16.1% Bell_Beaker_Czech + 83.9% Unetice_EBA» «2.5747»
2) файлы модели K11 и K16 для более сложной программы 4Admix (разработанной Александром Бурнашевом). Вторым инструментом вторичного анализа является 4Mix. Он работает по методу brute-force, шаг за шагом перебирая все возможные комбинации, а по окончанию цикла программа возвращает результат с наименьшим евклидовым расстоянием (по выбору можно использовать гауссово сглаживание, снижающее случайный статистический шум результатов). Как и в классическом Oracle, комбинация cмешиваемых этнических групп не может содержать более 4 популяций, хотя в отличие от классического Oracle, программа может моделировать комбинации из 3 и 4 этнических групп.
Пример. Приведу пример этих 3- и 4-членных аппроксимаций. В принципе, все то же самое, c той лишь разницей что теперь программа выделяет в комбинациях балтийскую и славянскую составляющую. Интересно, что скандинавская составляющая никуда не исчезла, оставаясь в пределах 20-25%
Using 3 populations approximation:
1 50% Belarusian_EastBelarus +25% English_Kent_GBR +25% Latvian @ 0.973956
2 50% Belarusian_EastBelarus +25% English_Kent_GBR +25% Lithuanian @ 0.988467
3 50% Latvian +25% French +25% Balt @ 1.036492
4 50% Lithuanian_Zemajitia +25% French +25% Irish_Connacht @ 1.05259
5 50% Lithuanian +25% Sorb +25% French_West @ 1.059638
6 50% Belarusian +25% Icelandic +25% French_West @ 1.06158
7 50% Lithuanian_Zemajitia +25% French +25% Irish_Cork_Kerry @ 1.074796
8 50% Lithuanian_Aukstajtia +25% French_East +25% Irish_Connacht @ 1.076771
9 50% Lithuanian_Zemajitia +25% French +25% Irish_Ireland @ 1.078576
10 50% Belarusian +25% Norwegian +25% French_West @ 1.079741
11 50% European_Utah +25% Lithuanian_Zemajitia +25% Balt @ 1.084317
12 50% Dane +25% Belarusian_EastBelarus +25% Lithuanian_Aukstajtia @ 1.090086
13 50% Lithuanian_Zemajitia +25% French +25% Scottish_Highlands @ 1.093951
14 50% Lithuanian +25% North_European +25% Sorb @ 1.103744
15 50% Lithuanian_Aukstajtia +25% English_GBR +25% French_Northwest @ 1.105369
16 50% Lithuanian_Zemajitia +25% French +25% Scottish_Grampian @ 1.106616
17 50% Lithuanian_Aukstajtia +25% French_Northwest +25% Irish_Connacht @ 1.106771
18 50% Lithuanian_Aukstajtia +25% French_Northwest +25% Scottish_Dumfries_Galloway @ 1.108261
19 50% Lithuanian +25% French_West +25% Polish_West @ 1.113695
20 50% Latvian +25% North_European +25% Sorb @ 1.115164
31501779 iterations.
Using 4 populations approximation:
1Belarusian_EastBelarus+Lithuanian_Zemajitia+Swede+French_West @ 0.947002
2Belarusian_EastBelarus+English_Kent_GBR+Lithuanian_Aukstajtia+Sorb @ 0.971605
3Belarusian_EastBelarus+Belarusian_EastBelarus+English_Kent_GBR+Latvian @ 0.973956
4Belarusian_EastBelarus+English_Kent_GBR+Lithuanian_Aukstajtia+Polish_East @ 0.986863
5Belarusian_EastBelarus+Belarusian_EastBelarus+English_Kent_GBR+Lithuanian @ 0.988467
6 French+Lithuanian_Zemajitia+Swede+Balt @ 0.98916
7Belarusian_EastBelarus+English_Kent_GBR+Lithuanian_Aukstajtia+Polish @ 0.996302
8 Belarusian+Lithuanian_Aukstajtia+Shetlandic+French_West @ 1.010485
9 Belarusian+Lithuanian_Zemajitia+Irish_Ulster+French_West @ 1.01227
10 Belarusian+Lithuanian_Zemajitia+French_West+Irish_Ulster @ 1.012977
11 Belarusian_EastBelarus+Lithuanian_Aukstajtia+Swede+Welsh @ 1.013043
12Belarusian_EastBelarus+European_Utah+Lithuanian_Aukstajtia+Swede @ 1.013805
13Belarusian_EastBelarus+Lithuanian_Aukstajtia+Swede+French_West @ 1.018296
14German_NorthGermany+Lithuanian_Aukstajtia+Balt+French_West @ 1.026503
15 Lithuanian_Aukstajtia+Sorb+Ukrainian-Center+French_West @ 1.027473
16 Belarusian+Lithuanian_Zemajitia+French_West+Irish_Connacht @ 1.031967
17Belarusian+Lithuanian_Zemajitia+French_West+Irish_Cork_Kerry @ 1.035716
18 French+Latvian+Latvian+Balt @ 1.036492
и т.д.
То же самое, но в модели K11
Using 3 populations approximation:
1 50% Poltavka_MBA_outlier +25% Halberstadt_LBA +25% Hungary_BA @ 2.031302
2 50% Poltavka_MBA_outlier +25% Bell_Beaker_Czech +25% Hungary_BA @ 2.072453
3 50% British_AngloSaxon +25% Halberstadt_LBA +25% Poltavka_MBA_outlier @ 2.125791
4 50% Bell_Beaker +25% Bell_Beaker +25% LesCloseaux13_Mesolithic @ 2.209118
5 50% Halberstadt_LBA +25% British_AngloSaxon +25% Poltavka_MBA_outlier @ 2.244371
6 50% Halberstadt_LBA +25% Hungary_BA +25% Samara_HG @ 2.270667
7 50% Halberstadt_LBA +25% Poltavka_MBA_outlier +25% Unetice_EBA @ 2.291406
8 50% Poltavka_MBA_outlier +25% British_AngloSaxon +25% Hungary_BA @ 2.30791
9 50% Bell_Beaker_Czech +25% Hungary_BA +25% Samara_HG @ 2.356281
10 50% Halberstadt_LBA +25% Nordic_BA +25% Poltavka_MBA_outlier @ 2.358744
11 50% Bell_Beaker +25% Hungary_BA +25% Karelia_HG @ 2.369978
12 50% Bell_Beaker_Czech +25% Nordic_BA +25% Poltavka_MBA_outlier @ 2.385823
13 50% Halberstadt_LBA +25% Corded_Ware_Germany +25% Nordic_BA @ 2.490915
14 50% Poltavka_MBA_outlier +25% Hungary_BA +25% Unetice_EBA @ 2.503754
15 50% British_AngloSaxon +25% Bell_Beaker_Czech +25% Poltavka_MBA_outlier @ 2.53217
16 50% Halberstadt_LBA +25% Baltic_LBA +25% Halberstadt_LBA @ 2.540751
17 50% Hungary_BA +25% Poltavka_MBA_outlier +25% Samara_HG @ 2.551414
18 50% Poltavka_MBA_outlier +25% Alberstedt_LN +25% Hungary_BA @ 2.561557
19 50% British_AngloSaxon +25% Poltavka_MBA_outlier +25% Unetice_EBA @ 2.575398
20 50% Bell_Beaker_Czech +25% British_AngloSaxon +25% Poltavka_MBA_outlier @ 2.575919
1127348 iterations.
Using 4 populations approximation:
1 Halberstadt_LBA+Hungary_BA+Poltavka_MBA_outlier+Poltavka_MBA_outlier @ 2.031302
2 Halberstadt_LBA+Nordic_BA+Poltavka_MBA_outlier+Unetice_EBA @ 2.03713
3 Bell_Beaker_Czech+Hungary_BA+Poltavka_MBA_outlier+Poltavka_MBA_outlier @ 2.072453
4 British_AngloSaxon+Halberstadt_LBA+Poltavka_MBA_outlier+Unetice_EBA @ 2.088049
5 British_AngloSaxon+British_AngloSaxon+Halberstadt_LBA+Poltavka_MBA_outlier @ 2.125791
6 British_AngloSaxon+Halberstadt_LBA+Hungary_BA+Samara_HG @ 2.131526
7 Bell_Beaker_Czech+Halberstadt_LBA+Hungary_BA+Samara_HG @ 2.14648
8 Bell_Beaker+Bell_Beaker+Bell_Beaker+LesCloseaux13_Mesolithic @ 2.209118
9 Bell_Beaker_Czech+Halberstadt_LBA+Nordic_BA+Poltavka_MBA_outlier @ 2.209365
10 Bell_Beaker_Germany+British_AngloSaxon+Hungary_BA+Samara_HG @ 2.212982
11 Bell_Beaker_Czech+Bell_Beaker_Germany+Hungary_BA+Samara_HG @ 2.232922
12 British_AngloSaxon+Halberstadt_LBA+Halberstadt_LBA+Poltavka_MBA_outlier @ 2.244371
13 British_AngloSaxon+Halberstadt_LBA+Nordic_BA+Poltavka_MBA_outlier @ 2.254756
14 Alberstedt_LN+British_AngloSaxon+Hungary_BA+Samara_HG @ 2.255589
15 Bell_Beaker_Czech+British_AngloSaxon+Halberstadt_LBA+Poltavka_MBA_outlier @ 2.256027
16 Halberstadt_LBA+Halberstadt_LBA+Hungary_BA+Samara_HG @ 2.270667
3) новым инструментом в релизе будет R программа nMonte, разработанная голландцем Гером Гизбертом. В отличие от двух предыдущих инструментов (ограниченных в числе используемых для моделирования этнических групп), nMonte позволяет использовать для моделирования (аппроксимации) генмоа все референсные грппы. Программа использует алгоритм эволюционного моделирования по методу Монте-Карло.
После пошагового добавления новой популяции программа определяет уменьшается ли евклидово расстояние; если да, то шаг сохраняется, в противном случае шаг отклоняется. Алгоритм завершает свою работу после выполнения примерно миллиона шагов. Как и два предыдущих инструмента программа стремится к минимализации евклидова расстония; но похоже за счет использования метода Монте-Карло, алгоритм гораздо более эффективен. И, также, как и в других инструментах, в nMonte «наилучшая комбинация» определяется как комбинация с наименьшим расстоянием. Недостаток же nMonte состоит в том, что она выдает только наилучшее подходящее решение, в то время как Oracle представляет альтернативные варианты.
Пример. Посмотрим, сколько потенциальных предковых популяций выдаст nMonte при аппроксимации моего генома.
При первом запуске программа выдала комбинацию (в cкобках процентный вклад референсной популяции) следующих 65 популяций. Также как и в других инструментах, тон задают балтийские популяции, а также белорусы, сорбы и поляки.
Lithuanian_Zemajitia 10.1
Latvian 7.85
Lithuanian_Aukstajtia 7.85
Belarusian_SouthBelarus 6.55
Lithuanian 6.5
Pole 5.45
Belarusian_WestBelarus 4.8
Balt 4.35
Sorb 3.35
Belarusian 3.05
Belgian 3
Norwegian 2.95
Czech 2.75
Dane 2.5
Slovak 2.4
Icelandic 1.9
Swede 1.9
French_SouthFrance 1.5
Slovenian 1.5
Basque_Spanish 1.3
Frisian 1.15
German_NorthGermany 1.1
Sardinian 1.1
Polish_East 1.05
Ukrainian_WestUkraina 1
Polish 0.95
Basque_French 0.9
Orcadian 0.7
Spanish_Pais_Vasco_IBS 0.7
Hungarian 0.65
Irish_Connacht 0.65
DonKuban_cossack 0.6
Dutch 0.6
Ukrainian_EastUkraina 0.6
Scottish_Argyll_Bute_GBR 0.55
European_Utah 0.5
English_GBR 0.45
Croatian 0.4
Russian-Pskov 0.4
French_South 0.4
Welsh 0.35
Irish_Ulster 0.35
Scottish_Fife 0.3
German_SouthGermany 0.25
Scottish_Dumfries_Galloway 0.25
Belarusian_CentralBelarus 0.2
Datog 0.2
English_Cornwall_GBR 0.2
North_European 0.2
Ukrainian 0.2
Russian_Orjol 0.15
Afar 0.1
Belarusian_EastBelarus 0.1
English_Kent_GBR 0.1
Irish 0.1
Kambera 0.1
Russian_Smolensk 0.1
Vindija 0.1
Belarusian-East 0.1
Spanish_Canarias_IBS 0.1
Spanish_Cantabria_IBS 0.1
Spanish_Cataluna_IBS 0.1
Peruvian 0.05
Russian_Voronezh 0.05
В K11 показаны следующие палеогеномы (или их группы). По-прежнему, основа генома 40% моделируется как геном представителя культуры колоколовидных кубков.
«Bell_Beaker» 40.3
«Halberstadt_LBA» 31.6
«Samara_HG» 8.5
«Tyrolean_Iceman_EN» 2.05
«Esperstedt_MN» 1.95
«Swedish_Mesolithic» 1.95
«BerryAuBac_Mesolithic» 1.85
«Swedish_Motala_Mesolithic» 1.7
«Bichon_Azillian» 1.6
«Continenza_Paleolithic» 1.5
«Hungary_BA» 1.5
«LaBrana_Mesolithic» 1.35
«Bell_Beaker_Germany» 1.05
«Hungary_HG» 0.85
4) следующим новым инструментом будет 4mix, более упрощенный вариант 4Admix. Он разработан тем же Г. Гизбертом. Основное отличие от 4Admix — если 4Admix перебирает все возможные комбинации из 4 популяций, то в 4mix можно эксплицитно задавать отдельные комбинации и определять евклидову дистанции между этой комбинацией и аппроксимируемым геномом в пространстве моделей
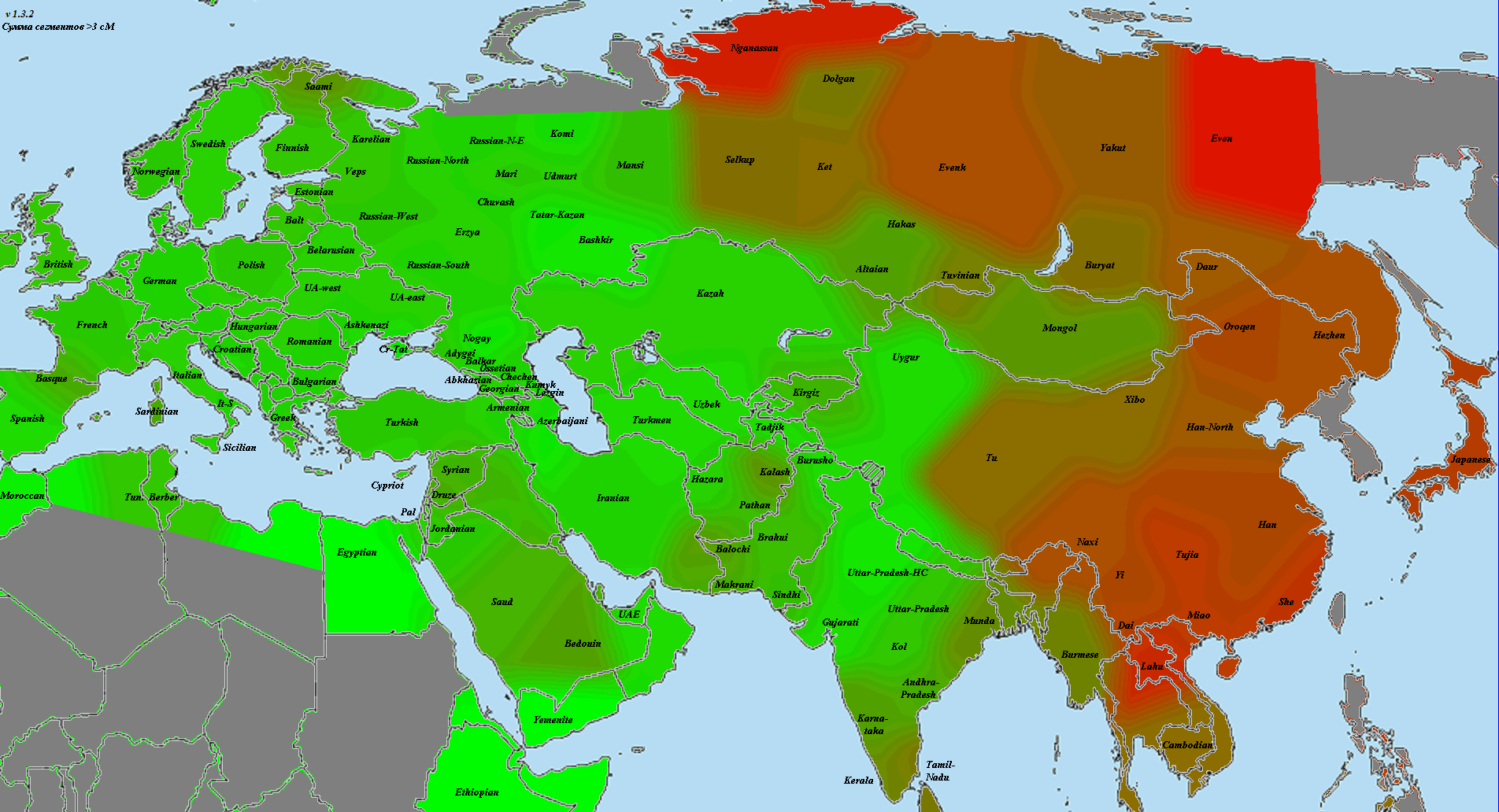
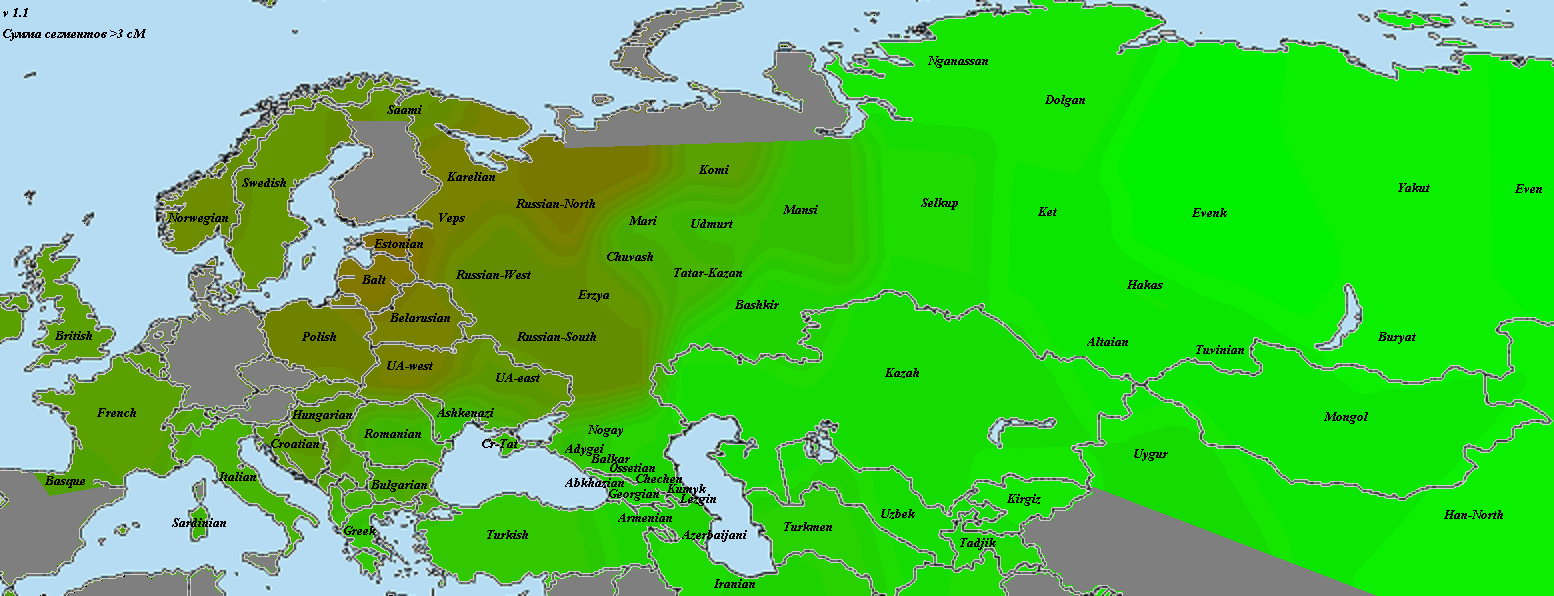
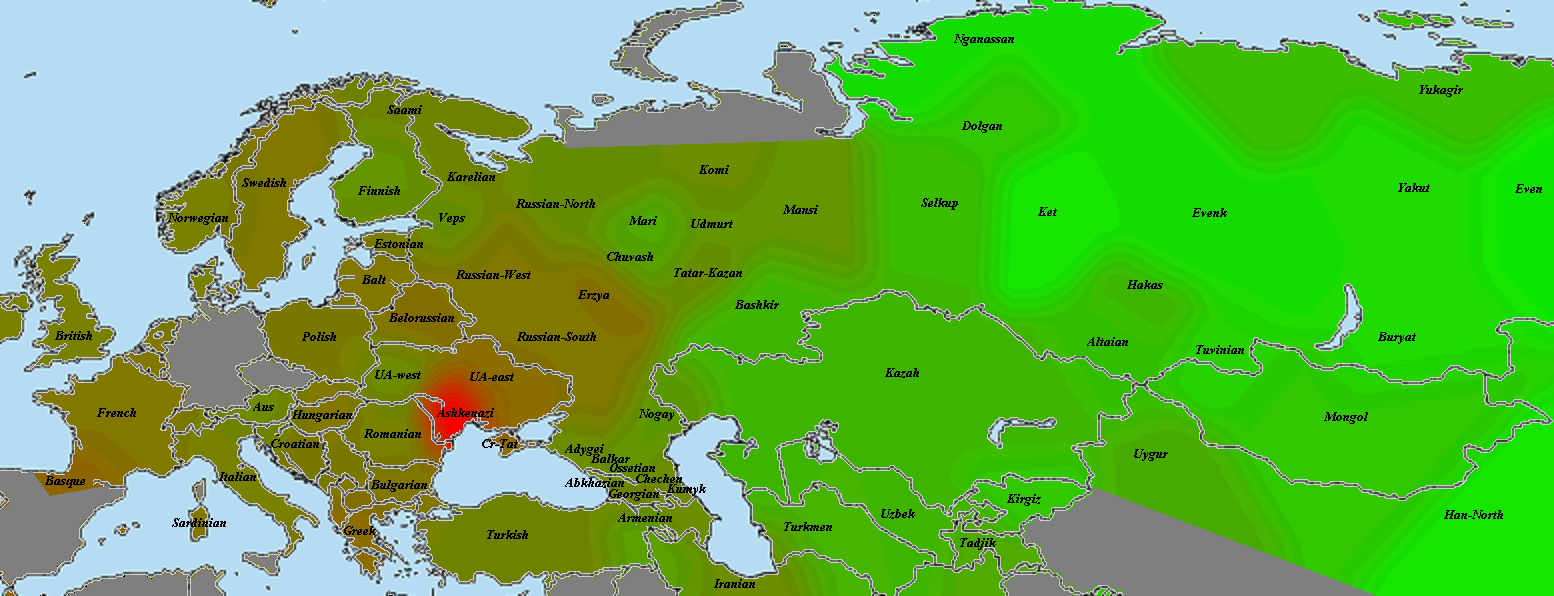
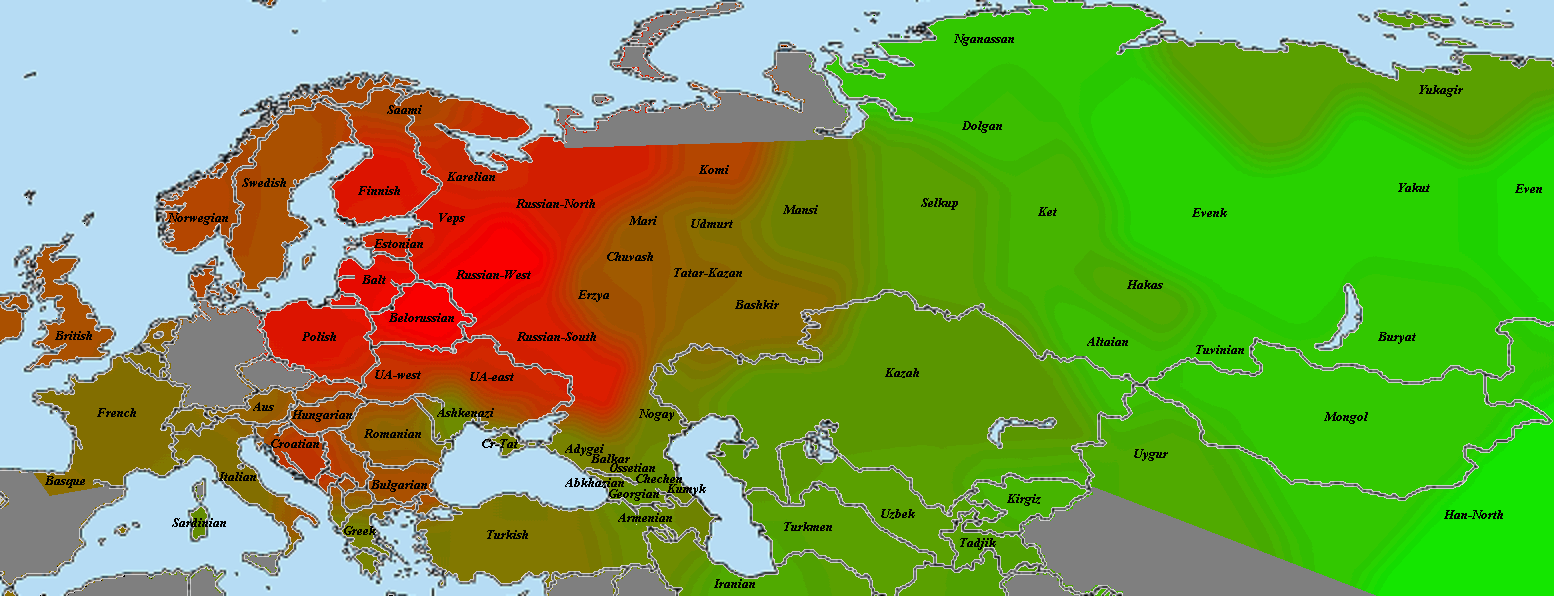
5) карты компонентов с аннотацией. Аннотации компонентов будут чуть позже, а вот карты уже готовы
Карты распространения некоторых компонентов K16 и K11 в ряде географических ареалов


















6) я включил в релиз модифицированный скрип GPS лаборатории Элхайка для определения географического ареала происхождения предков человека, чей геном является предметом изучения. Я включил пару строчек кода для проецирования вычисленных географических координат на географическую карту.
Пример. Ниже показаны две карты, на которые спроецированы географические координаты вычисленной алгоритмом GPS (GPS DNA tool ) точки «этнического происхождения».
Я проверил работоспособность алгоритма на обеих моделях.
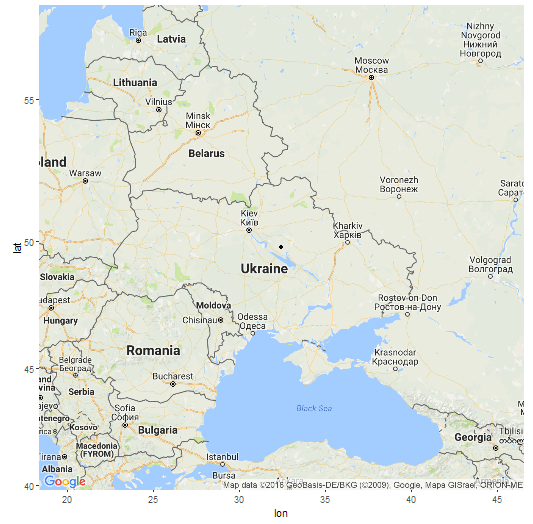
В модели K16 (современные популяции) GPS-координаты точки моего «происхождения» 49.7648663288835 32.4345922625112 (примерно 49 градусов северной широты и 32 градуса восточной долготы), т.е где-то на левом берегу Днепра в Украине. Как утверждают разработчики программы, она позволяет определить место происхождения с радиусом погрешности в 500 км. Я вычислил расстояние от полученной точки до настоящего места жительства предков (южная часть Брестской области) и получилось 470 км. Т.е точка попадает в радиус, хотя и с некоторым трудом.
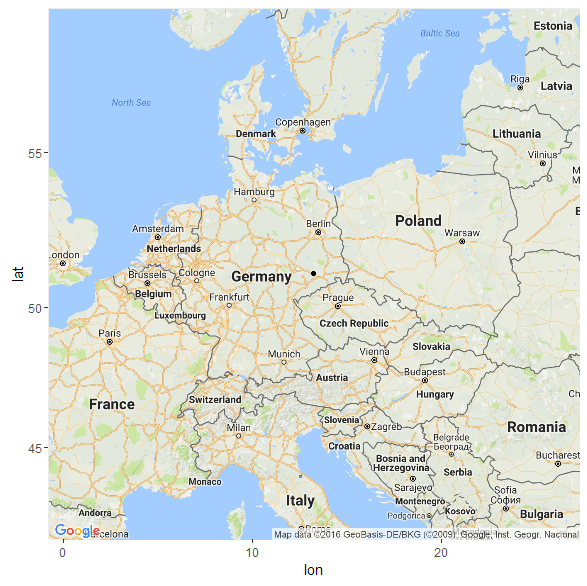
Что касается модели K11 (древние геномы), то в этой модели мой «Urheimat» локализуется — весьма ожидаемо — на землях древней унетицкой и лужицких культур (51.1254133094371 13.2336209988448)


























































Для отправки комментария необходимо войти на сайт.