Поскольку слово «экзом» является совсем свежым заимствованием из английского языка. Наиболее простое определеие: экзом состоит из совокупности экзонов, а экзон — это участок гена (ДНК) эукариот, несущий генетическую информацию, кодирующую синтез продукта гена (белка). Соответствующие экзонам участки ДНК, в отличие от интронов, полностью представлены в молекуле информационной РНК, кодирующей первичную структуру белка. По мнению некоторых исследователей соответствуют доменам (структурно автономным областям) в белке и являются первичными генетическими единицами, рекомбинация которых приводит к возникновению в ходе эволюции новых генов и соответственно новых белков. Э. чередуются в структуре гена с другими фрагментами — интронами.
Иными словами, экзом — это совокупность всех участков ДНК, несущих информацию, определяющую экспрессию белка.
Здесь уместно вспомнить недавную видео-лекцию Павла Певзнера «Персональная медицина и ассемблирование геномов: паззл с миллиардом частей» (где-то на мордокніге я давал ссылку). Певзнер, в числе прочего, мимоходом упоминул про недавную работу одного из ведущих сотрудников института Сангера (ведущего центра персональной геномики, одним из исследовательских направлений как раз и является -The 500 Exome Project with collaborators from WTSI, GSK and Lausanne University ).
Речь идет о нашумевшей работе, в котором ученый описывает как на протяжении полугода он в рутинном режиме ежемесячно «проверял экзомы» на предмет анализа экспрессии белков. В ходе работы был не только выявлен целый ряд ранее неизвестных вариантов генов, ответственных за предрасположенность к диабету второго типа, но и произведен анализ динамики белковых изменений.Этот тщательный анализ позволил «излечить» пациентов от диабета.
После того как медиа взбудоражила общественное сознание этой новостью, целая группа коммерческих компаний обратилась к этому, ранее коммерчески неосвоенному типу генотипирования (хотя некоммерческіе исследования ведутся уже не менее десятка лет). Чутко следящая за коньюктурой геномного рынка компания Illumina сразу опустила планку цен на «экзомы» до 200 долларов. Чем не приминула воспользоваться компания 23andme, предлагающая (в качестве посредника, т.к. само типирование проводится в лабах Иллюмины) конечному потребителю продукт по цене 999 долларов.Легкая доступность экзомного тестирования будет иметь свои преимущества, поскольку позволит не только проводить анализ генетических маркеров, определяющие риски, но и анализировать экспрессию белков под воздействием определенных эпигенетических факторов (тип приминяемых медикаментов, питания и т.д.).
Сейчас врачи посылают пациентов на анализ крови, cлюны, мочи и прочих биологических субстанций.Лет этак через 10 врач будет писать в истории болезнии: «Пациенту назначено прохождение годового курса экзомного генотипирования и анализа»
Перед тем как перейти к конкретному примеру, немного сухой теории.
Принципы и платформы экзомного сивенирования
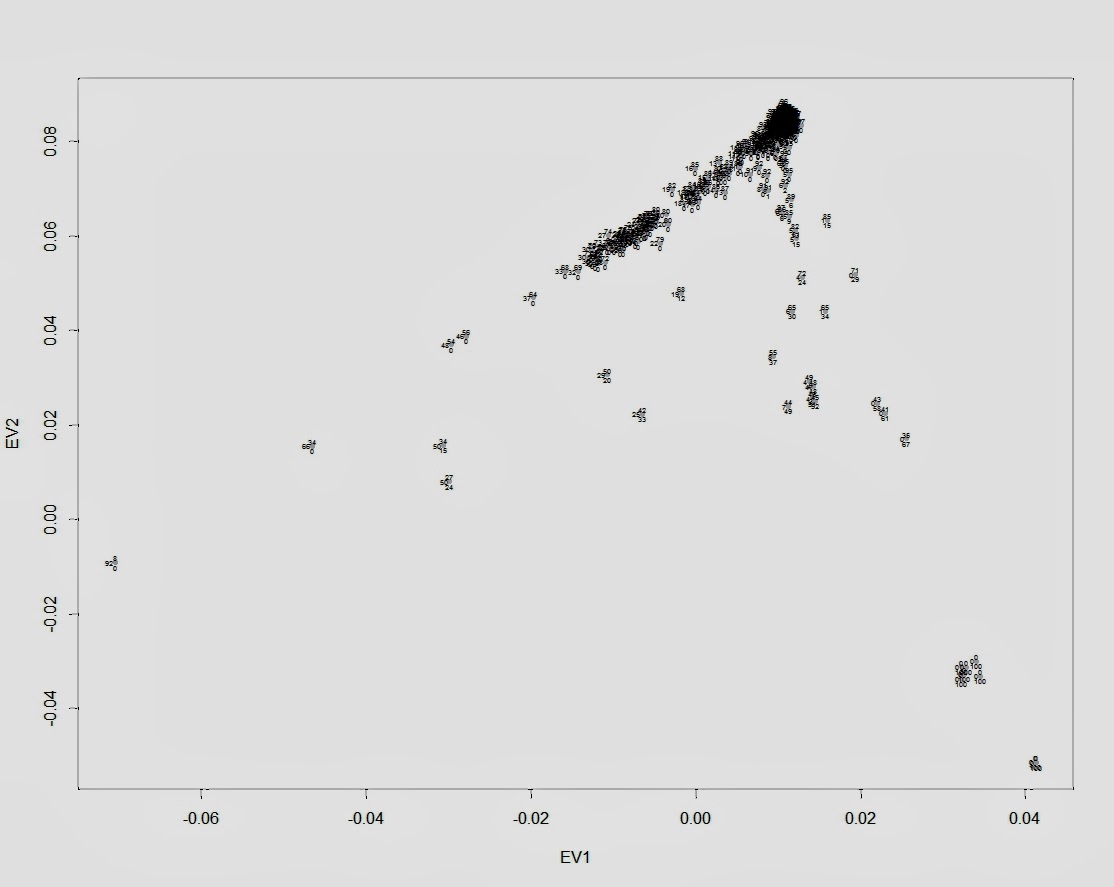
Авторы ряда недавних исследований пытались сравнить коммерческие технологии экзомного секвенирования. Предложенные в течение последних нескольких лет коммерческие платформы секвенирования следующего поколения секвенирования, разработаы с целью секвенирования кодирующей части генома — так называемого экзома. В недавнем исследовании лаборатории Майкла Снайдера (Стэнфорд), авторы сравнили три основных платформы экзомного секвенирования созданные компаниями Agilent’s SureSelect Human All Exon (50 Mbp), Roche/Nimblegen’s SeqCap EZ v2.0, and Illumina’s TruSeq Exome Enrichment. Платформы сравнивались как между собой, так и с платформой полногеномного сиквенирования (35x) на примере сиквенирования генома одного и того же человека.

Рис.1. Основные экзомные платформы (Clark и др., Nat Biotech, 2011 год.).
Различия платформ
Для начала, следует отметить различия между платформами.

Фото: Кларк и др., Nat. Biotech. 2011
Большое количество базовых пар генома (29,45 Мбит), которое (предположительно) составляет «ядро» покрывается всеми сравниваемыми платформами. В индивидуальном плане, каждая из платформ имеет от 4 до 28 Мбит уникального «таргетного пространства покрытия». Платформа Agilent лучше подходит для транскриптов Ensembl, в то время как платформа NimbleGen имеет более широкий охват микроРНК. На приведенной выше диаграмме Венна видно что пересчение множеств покрываемых платморфмами базовых пар генома («таргетное пространство») гораздо больше, чем пересечение каждой из платформ с множеством таргентного пространства платформы Illumina. Это в первую очередь объясняется тем, что Illumina в первую очередь нацелена на секвенирование нетранслируемых областей генома (НТО). Трудно сказать, является ли это преимуществом платформаы Illumina, или нет. С одной стороны, этот факт, конечно же, представляет определенный интерес исследователям, которые ищут геномные варианты в регионах НТО. С другой стороны, эта характерная особенность экзомной платформы Illumina приводит к неизбежному ухудшению качества покрытия генома. И, действительно, как отмечают авторы отмечают, при секвенировании генома в режиме 50 миллионов 2 × 100 б.п. покрытие генома на платформе Illumina составляет 30x, для сравнения 60x на платформе Agilent -60x и на платформе NimbleGen — 68x.
Определение таргетной эффективности платформы и содержание GC
Авторы произвели определение и секвенирование экзома на одном образце — взятом у добровольца европейского происхождения — с использованием всех трех экзомных решений. Каждая экзомная библиотека получила одну полосу прочтения (2 × 100 б.п.) на Illumina HiSeq 2000 (11-18 Гигобаз в каждой библиотеке). С помощью программы BWA 99% полученных геномных «ридов» было отображено на референсную последовательность человеческого генома, примерно 10-15% геномных ридов оказались ПЦР-дубликатами. Затем была вычислена общая таргетная эффективность прочитки ( при расчете исходили из того, что на каждый экзом приходится примерно 80 миллионов ридов, и вычисляли процентные доли нуклеотидов покрытых при прочитке 10x, 20x, 30x ). Как пишут авторы исследования: «при всех аналогичных параметров покрытия и числа ридов, платформа NimbleGеn дала более высокий процент опеределения своих «»таргетных» (целевых) баз, в сравнении с другими платформами.» Авторы объясняют эффективность платформы использованием высокой плотности перекрывающихся биотинилированных олигов, используемых в платформе NimbleGen для «захвата» экзонов.
Неудивительно, что все платформы продемонстрировали заметное снижение охвата при повышении и понижении GC целей. Однако, при низкой GC (40% до 20%), платформа Agilent показал лишь небольшое cнижение глубины прочитки, что, возможно, связано с меньшим числом циклов ПЦР, и большим числом биолитинированных пробов для захвата РНК, а также с использованием уникальных РНК-пробов.
Обнаружение единичных нуклеотидных вариантов (SNV-ов) и небольших инделов
Как известно, обнаружение небольших вариантов последовательности, особенно SNV-ов, является одной из основных целей секвенирования экзома. Используя нормированные наборы геномных «ридов» размером в ~ 80 мегабаз, авторы обнаружили в каждом наборе экзома SNV-ы (авторы использовали программное обеспечения GATK). Все три платформы показали высокую степень корреляции между «вызовами» SNV-ов и высокой плотностью массива генотипов SNP-ов. Как показал эксперимент, референсный аллель имел небольшое преимущество (0.53-0.55) в позициях SNP-ов, и это дает повод утверждать о небольшом отклонении результатов в случае с ридами, которые содержат геномные варианты. Вместе с тем, не было найденно никаких отклонений в отношении определенных типов субституций. Как и следовало ожидать, на всех платформах количество обнаруженных SNV-ов увеличивалось при увеличении охвата генома. Этот рост, однако, не носил линейный характе; при прочитке 30 миллионов баз, не было обнаружено более 95% SNV-ов. В общих регионах секвенированных экзомов, платформа NimbleGen обнаружила большинство SNVs при наименьшем количестве прочтений.
Кроме того, платформа NimbleGen (за счет более эффективного захвата и, следовательно, более глубокого охвата генома) обнаружила большинство инделов — как в общих регионах экзомов, так и в регионах рефересной последовательности генома. При более низком уровне прочитке, платформа Agilent обнаружила больше инделов в общих регионах экзомов, но при 50 миллионах прочтений, платформа Illumina превзошла Agilent (и, что неудивительно, обнаружила еще много инделов в нетранслируемых регионах). Размер большинства инделов был равне 1 базовой паре, хотя авторы отметили небольшое увеличение размера инделов до 4-8 базовых пар (что подтверждается путем сравнения генома человека с геномом приматов), а также увеличение за счет отбора против сдвига рамки мутаций.
Сравнение результатом экзомного секвенирования с результатами полногеномного секвенирования
Основным достоинством данного исследования можно считать то, что авторы произвели и полногеномное секвенирование образца при средней величине покрытия 35х. Корреляция по гетерозиготным позициям SNP-ов между результатами экзомного секвенирования и полногенмного секвенирования составила 98,5%. Для имитации мультиплексного секвенирования 3 или 6 экзомных библиотек на одну полосу (GAIIx или HiSeq, соответственно), авторы нормировали экзомные продукту до 50 миллионов ридов на каждой из платформ. В каждом попарном сравнении продуктов полногеномного и экзомного секвкенирования, набор данных полногеномного секвенирования был ограничен теми же таргетными регионами, что и сравниваемый экзомный продукт. Хотя этот шаг и представляется необходимым для сравнения по методу «подобное к подобному», нужно отметить, что он сводит к минимуму мощь полногеномного секвенирование, которое обеспечивает относительно объективное освещение всех кодирующих областей. Другими словами, это ограничение дает определенное преимущество экзомным продуктам, поскольку сравниваются только те таргетные регионы, под которые заточена платформа экзомного продукта.

Перекрытие SNV-ов в экзомных продуктах и полно-геномном продукте (Clark и др., Nat Biotech 2011 года.)
Подавляющее большинство SNV-ов в таргетных регионах было обнаружено как в экзомных, так и в полногеномных регионах, с небольшими различиями. Примечательно, что при попарном сравнении специфические SNV в экзомных и полногеномных продуктах, ка правило, имеют (1) низкий доверительный порог, (2) более высокую долю новельных (по отношению к dbSNP) вариантов, и (3) лучшее покрытие в детектирующей платформе. Специфические для полногеномного продукта SNV-ы часто имеют нулевые риды в экзомных продуктах (вероятно, из-за проблемы с гибридизацией). Напротив, большинство SNV-ов, специфических для экзомных продуктов, было покрыто в полногеномном продукте, хотя их общее число в полногеномном продукте все равно остается ниже, чем в экзомном продукте.
Как становится ясно из вышеприведенного рисунка, число SNV-ов, обнаруженых в экзомных продуктах и полногеномном продукте отлично коррелирует с «»потолком» каждой экзомной платформы. Illumina, которая имеет наибольшее таргетное пространство (особенно в нетранслируемых регионах), имеет наибольшее количество общих SNV-оd. Число общих SNV-ов в Agilent больше, чем NimbleGen, однако чувствительность NimbleGen в определении истинно-положительных результатов в целевых регионах гораздо выше, чем на двух других платформах.
Как выбрать экзомную платформу
Авторы приходят к выводу, что все три платформы экзомного секвенирования по-своему хороши. Выбор платформы, вероятно, зависит от целей, приоритетов и бюджета исследователя. Для малобюджетных проектов, NimbleGen предлагает наиболее эффективное обогащение экзонов (а также микро-РНК). Agilent подходит для охоты за вариантами генома, поскольку обеспечивает более широкий охват, но требует больше секвенированных данных. Illumina наиболее требовательна в плане секвенированных даннных, но зато обследует нетранслируемые области, могут заинтересовать некоторых исследователей.
Мои собственные практические выводы на основе анализа данных
Летом я освоил навыки анализоа результатов экзомного генотипирования, любезно представленные одним из немногих россиян, участвовавших в пилотном проекте экзомного генотипирования в компании 23andme.
Поскольку слово «экзом» является совсем свежым заимствованием из английского языка. Наиболее простое определеие: экзом состоит из совокупности экзонов, а экзон — это участок гена (ДНК) эукариот, несущий генетическую информацию, кодирующую синтез продукта гена (белка). Соответствующие экзонам участки ДНК, в отличие от интронов, полностью представлены в молекуле информационной РНК, кодирующей первичную структуру белка. По мнению некоторых исследователей Э. соответствуют доменам (структурно автономным областям) в белке и являются первичными генетическими единицами, рекомбинация которых приводит к возникновению в ходе эволюции новых генов и соответственно новых белков. Э. чередуются в структуре гена с другими фрагментами — интронами.
Иными словами, экзом — это совокупность всех участков ДНК, несущих информацию, определяющую экспрессию белка.
Здесь уместно вспомнить недавную видео-лекцию Павла Певзнера «Персональная медицина и ассемблирование геномов: паззл с миллиардом частей» (где-то на мордокніге я давал ссылку). Певзнер, в числе прочего, мимоходом упоминул про недавную работу одного из ведущих сотрудников института Сангера (ведущего центра персональной геномики, одним из исследовательских направлений как раз и является -The 500 Exome Project with collaborators from WTSI, GSK and Lausanne University ).
Речь идет о нашумевшей работе, в котором ученый описывает как на протяжении полугода он в рутинном режиме ежемесячно «проверял экзомы» на предмет анализа экспрессии белков. В ходе работы был не только выявлен целый ряд ранее неизвестных вариантов генов, ответственных за предрасположенность к диабету второго типа, но и произведен анализ динамики белковых изменений.
Этот тщательный анализ позволил «излечить» пациентов от диабета.
После того как медиа взбудоражила общественное сознание этой новостью, целая группа коммерческих компаний обратилась к этому, ранее коммерчески неосвоенному типу генотипирования (хотя некоммерческіе исследования ведутся уже не менее десятка лет). Чутко следящая за коньюктурой геномного рынка компания Illumina сразу опустила планку цен на «экзомы» до 200 долларов. Чем не приминула воспользоваться компания 23andme, предлагающая (в качестве посредника, т.к. само типирование проводится в лабах Иллюмины) конечному потребителю продукт по цене 999 долларов.
Легкая доступность экзомного тестирования будет иметь свои преимущества, поскольку позволит не только проводить анализ генетических маркеров, определяющие риски, но и анализировать экспрессию белков под воздействием определенных эпигенетических факторов (тип приминяемых медикаментов, питания и т.д.).
Сейчас врачи посылают пациентов на анализ крови, cлюны, мочи и прочих биологических субстанций.
Лет этак через 10 врач будет писать в истории болезнии: «Пациенту назначено прохождение годового курса экзомного генотипирования и анализа»
Как выглядит конечный продукт экзомного тестирования предлагаемый 23andme за 999 зеленых американских рублей?
Это набор из четырех файлов:
1) x.bam
2) x.bai
3) x.pdf
4) x.vcf.
X -это кодовый номер участника. BAM файл являющийся бинарной версией формата SAM (формата множественного выравнивания ДНК по референсному сиквенсу), BAI — индекс контигов в BAM файле. Наконец, VCF — это файл содержащий все «задетектированные» в BAM файле варианты (прежде всего SNPs и INDELs)
Но вернемся к экзомному тестированию.
Cуществует определенная группа лиц, которых больше интересуют вопросы происхождения и генеалогии. Медицинские аспекты, как правило, им неинтересны.
Что интересного могут излечь из экзомных данных ДНК-генеалогии? Не трудно ответить. Большинство ДНК-генеалогов знает принципы наследования ДНК, в первую очередь Y-хромосомы и митохондриального генома (которые наследуются соответственно строго по мужской и женской линии).
После предварительного знакомства со структурой экзомных данных, я должен выделить две основные проблемы, возникающие при работе с указанными выше «однородительскими маркерами».
Первая и основная проблема — это характер экзомного типировния. При экзомном типировании определяются только те снипы и инделы, которые находятся в экзонах. С митохондрионом здесь проблем особых нет — в [человеческом] митогеноме практически все вариативные позиции, являются экзомными, т.е несут генетическую информацию («код» синтеза белка). Поэтому фактически данные экзомного тестирования уже содержат полный сиквенс генома (аналог FGS от FTDNA). Остается только их извлечь. И вот тут появляется другая проблема. Для определения генетических вариантов (т.е различий нуклеотидов в локусах) необходмо провести «выравнивание» анализируемого по референсному сиквенсу. Как известно, в митохондрионе для этих целей используется «классический» сиквенс rCRS (Cambridge Reference Sequence, GenBank:NC_012920.1). Однако в геномных билдах-ассамблеях hg18 и hg19_Chr37, этот референс заменен другим. Поэтому результаты выравнивания митогенома по дефольтным вариантам вышеуказанных билдов дают результаты, сильно отличающиеся от привычного формата.
После замены дефолтного сиквенса на rCRS все получилось. Вот фрагмент из VCF файла, содержащий интересующие нас отличия от rCRS:
#CHROM POS REF ALT
MT 73 A G
MT 195 T C
MT 263 A G
MT 709 G A
MT 750 A G
MT 1438 A G
MT 1888 G A
MT 2141 T C
MT 2706 A G
MT 3106 CN C
MT 4216 T C
MT 4917 A G
MT 5894 A G
MT 7028 C T
MT 8697 G A
MT 8860 A G
MT 9117 T C
MT 10463 T C
MT 11191 C T
MT 11251 A G
MT 11719 G A
MT 11812 A G
MT 12741 C T
MT 13260 T C
MT 13368 G A
MT 13965 T C
MT 13966 A G
MT 14233 A G
MT 14687 A G
MT 14766 C T
MT 14905 G A
MT 15326 A G
MT 15452 C A
MT 15607 A G
MT 15928 G A
MT 16126 T C
MT 16294 C T
MT 16296 C T
MT 16324 T C
MT 16519 T C
Теперь о Y-хромосоме. В отличии от митохондриона, где практически все снипы локализуются в экзонах, больша часть снипов мужской Y-хромосомы лежит в «информационно бесполезных» интроных зонах. Поскольку экзомное тестирование не покрывает интроны, то большинство из известных Y-снипов просто выйдет за рамки теста
Убедился и я в этом на примере реальных данных (это представитель Y хромосомной гаплогруппы R1a1).
samtools view -h x.bam Y > Y.sam
samtools view -h -b -S Y.sam > Y.bam
samtools/samtools mpileup -C 50 -ugf chrY.fa Y.bam | /samtools/bcftools/bcftools view -vcg — > Y.raw.vcf
Данный подход позволил обнаружить у тестанта около сотни генетических полиморфизмов (координаты данные по билду hg19):
Y 4058546 0 A C
Y 4058566 0 ta t
Y 4457069 0 tctctcct tct
Y 6028350 0 A T
Y 8149348 0 G A
Y 8566853 0 GCCC GCCCC
Y 8783761 0 C T
Y 8881927 0 GGTGT GGTGTGT
Y 9198243 0 T A
Y 9304866 0 G A
Y 9368340 0 tg tGNg
Y 9384631 0 A C
Y 9385720 0 CGG CG
Y 9909058 0 T A
Y 9930114 0 C A
Y 9931330 0 T A
Y 9938790 0 C A
Y 9938851 0 A T
Y 9938982 0 T C
Y 9939117 0 T A
Y 9952497 0 A G
Y 9982892 0 G A
Y 9982917 0 C A
Y 10007709 0 C A
Y 10007727 0 G A
Y 10007741 0 G A
Y 10011344 0 A G
Y 10011487 0 A G
Y 10011498 0 G C
Y 10011502 0 A G
Y 10011545 0 T G
Y 10011604 0 C CTT
Y 10011648 0 T G
Y 10011673 0 G A
Y 10011677 0 G A
Y 10011698 0 A G
Y 10011878 0 G A
Y 10011935 0 C CT
Y 10011960 0 T C
Y 10011966 0 ATT AT
Y 10012012 0 T A
Y 10013318 0 A G
Y 10028123 0 C T
Y 10028180 0 A G
Y 10029163 0 A G
Y 10029228 0 G A
Y 10029308 0 A T
Y 10029322 0 T C
Y 10029340 0 T C
Y 10029485 0 G C
Y 10029487 0 T A
Y 10029513 0 A G
Y 10029610 0 G A
Y 10029616 0 G T
Y 10029623 0 C T
Y 10029629 0 A G
Y 10029649 0 C G
Y 10029711 0 A C
Y 10043269 0 C T
Y 13241432 0 G T
Y 13241656 0 G A
Y 13243050 0 C G
Y 13243352 0 G A
Y 13244666 0 C T
Y 13244690 0 A G
Y 13254228 0 C T
Y 13262943 0 ACCC ACC
Y 13263091 0 G A
Y 13263304 0 C T
Y 13263364 0 A G
Y 13263374 0 C G
Y 13266266 0 G A
Y 13266286 0 C T
Y 13266301 0 A G
Y 13266368 0 T G
Y 13266377 0 G C
Y 13266499 0 A G
Y 13266520 0 G T
Y 13266556 0 T G
Y 13266560 0 C T
Y 13266587 0 C G
Y 13268187 0 T C
Y 13268361 0 T C
Y 13268377 0 A G
Y 13268521 0 C T
Y 13307425 0 G T
Y 13307562 0 G A
Y 13309174 0 A T
Y 13309226 0 A C
Y 13309239 0 G C
Y 13309262 0 T C
Y 13309348 0 C T
Y 13311223 0 T A
Y 13311491 0 C T
Y 13311501 0 G A
Y 13312579 0 G A
Y 13312666 0 G C
Y 13312729 0 C T
Y 13312756 0 A G
Y 13312789 0 A G
Y 13332277 0 C T
Y 13357224 0 C T
Y 13370991 0 C A
Y 13445929 0 G C
Y 13445957 0 C G
Y 13463779 0 A C
Y 13463831 0 T A
Y 13463837 0 G A
Y 13463860 0 C G
Y 13465055 0 A G
Y 13470805 0 G A
Y 13470834 0 T C
Y 13470855 0 T G
Y 13470880 0 G A
Y 13470897 0 G A
Y 13475849 0 C T
Y 13476553 0 T C
Y 13478387 0 A T
Y 13478445 0 G C,A
Y 13478569 0 T G
Y 13478583 0 T G
Y 13478613 0 A G
Y 13485671 0 T G
Y 13488312 0 C A
Y 13488330 0 A G
Y 13488337 0 C T
Y 13488370 0 G A
Y 13488395 0 A G
Y 13488410 0 A T
Y 13488429 0 A G
Y 13488601 0 A C
Y 13488621 0 A G
Y 13488946 0 A C
Y 13488952 0 T C
Y 13488972 0 C G,T,A
Y 13488988 0 A G
Y 13488992 0 T C
Y 13489043 0 G A
Y 13489069 0 A C,G
Y 13489077 0 T C
Y 13489206 0 C G
Y 13489220 0 T C
Y 13489234 0 T C
Y 13489255 0 A G
Y 13489292 0 A G
Y 13489300 0 A G
Y 13492264 0 C A
Y 13500410 0 T G
Y 13500424 0 T C
Y 13500443 0 T C
Y 13502048 0 C T
Y 13524378 0 T C
Y 13524752 0 G T
Y 13524761 0 C T
Y 13524873 0 T C
Y 13537129 0 G A
Y 13537569 0 A T
Y 13537581 0 C T
Y 13541022 0 C A
Y 13541053 0 CA CATA
Y 13541068 0 T C
Y 13541199 0 A G
Y 13541232 0 A T
Y 13541288 0 G A
Y 13541293 0 ATTT ATT
Y 13541420 0 A C
Y 13541454 0 T C
Y 13541478 0 G T
Y 13541520 0 C T
Y 13541556 0 A C
Y 13541561 0 T G
Y 13541584 0 C G
Y 13572922 0 A C
Y 13572932 0 T C
Y 13572999 0 A G
Y 13573033 0 A C
Y 13573108 0 G C
Y 13573152 0 C A
Y 13573216 0 G A
Y 13573240 0 C T
Y 13573271 0 G T
Y 13595280 0 T C
Y 13687807 0 T G
Y 13688825 0 C G
Y 13689634 0 T C
Y 13689668 0 C G
Y 13689755 0 G C
Y 13690562 0 C T
Y 13694899 0 G A
Y 13694929 0 G A
Y 13694956 0 C G
Y 13694983 0 T A
Y 13695051 0 T G
Y 13726074 0 T A
Y 13726129 0 C G
Y 13842718 0 G C
Y 14482235 0 C A
Y 14485120 0 G A
Y 14498990 0 C T
Y 14771478 0 A T
Y 14898094 0 A G
Y 14958218 0 C T
Y 15026424 0 A C
Y 15027529 0 T G
Y 15930958 0 ccttcttcctc cCTTCTTCCTCCTcttcttcctc
Y 16751825 0 A G
Y 16832517 0 T C
Y 17231616 0 A G
Y 21154004 0 A C
Y 21154323 0 G A
Y 21154426 0 G A
Y 21154466 0 T A
Y 21208056 0 A G
Y 21208066 0 C G
Y 22260237 0 C T
Y 22510104 0 G A
Y 22510163 0 T A
Y 23473201 0 T A
Y 23800360 0 T G
Y 23805478 0 C A
Y 24008079 0 T A
Y 28582510 0 G C
Y 28582566 0 C G
Y 28582605 0 T C
Y 28582622 0 G A
Y 28582676 0 G A
Y 28582685 0 C A
Y 28582863 0 A G
Y 28582865 0 A G
Y 28582921 0 A G
Y 28582932 0 G A
Y 28583310 0 C T
Y 28583314 0 A G
Y 28583382 0 G C
Y 28583394 0 T C
Y 28583410 0 C G
Y 28583415 0 T C
Y 28583431 0 A T
Y 28583432 0 A G
Y 28583590 0 A C
Y 28586782 0 G A
Y 28586959 0 T C
Y 28587232 0 T C
Y 28689055 0 G T
Y 28709343 0 A G
Y 28780767 0 A C
Y 28780823 0 T A
Y 28780883 0 G A
Y 28815270 0 C A
Y 28815656 0 T C
Y 28816806 0 T C
Y 28816831 0 C T
Y 28816870 0 T G
Y 28816948 0 C G
Y 28817276 0 T G
Y 28817286 0 T G
Y 28817559 0 T G
Y 28817636 0 G A
Y 58856145 0 G C
Y 58883603 0 A T,C
Y 58883784 0 T A
Y 58883834 0 A T
Y 58893627 0 A T
Y 58968939 0 G A
Y 58975896 0 T C
Y 58981639 0 cctccactcca cCTCCActccactcca
Y 58982160 0 G T
Y 58982559 0 A C
Y 58982671 0 tcttccttc tcttc
Y 58985524 0 T G
Y 58996230 0 G A
Y 58996257 0 G T
Y 58999765 0 C T
Y 58999773 0 G A
Y 59001429 0 G A
Y 59001608 0 C T
Y 59001620 0 A C
Y 59001647 0 G A
Y 59001685 0 G C
Y 59001722 0 G A
Y 59001753 0 T C
Y 59001773 0 A C
Y 59001782 0 C A
Y 59001792 0 T C
Y 59001960 0 T A
Y 59002047 0 C G
Y 59002139 0 G T,A
Y 59005179 0 C A
Y 59010280 0 A G
Y 59015256 0 T A
Y 59017005 0 A G
Y 59017181 0 T A
Y 59017206 0 A G
Y 59017378 0 T G
Y 59017384 0 ag aGg
Y 59018341 0 C G
Y 59020728 0 A G
Y 59022718 0 A G
Y 59022723 0 C T
Y 59022734 0 C T
Y 59022768 0 A G
Y 59027525 0 A G
Y 59027700 0 A C
Y 59027882 0 T G
Y 59029728 0 C T
В продолжение о Y-хромосоме. Совершенно ясно, что большинство из снипов, обнаруженных у протестированного ранее не были известны, и поэтому отсутсвуют в официальном списке ISOGG.
C помощью незамысловатой комманды grep -f snps ISOGGsnps я нашел лист известных ISOGG-снипов, которые также присутствуют и в данных тестанта>
L146 R1a M420 rs17250535 21882589 23473201 T->A
L265 R1b1a2 rs9786882 8209348 8149348 A->G
L269 G 13467612 14958218 T->C
M173 R1 P241; Page29 rs2032624 13535818 15026424 A->C
M201 G rs2032636 13536923 15027529 G->T
M379 I2a2a2 rs2032636 13536923..13536924 15027529..15027530 GT->del
M420 R1a L146 rs17250535 21882589 23473201 T->A
P241 R1 M173; Page29 rs2032624 13535818 15026424 A->C
Page7 R1a1a1 rs34297606 13008998 14498990 C->T
Page29 R1 M173; P241 rs2032624 13535818 15026424 A->C
Page83 P rs35361051 13407488 14898094 A->G
Исходя из вышеприведенной таблицы, Y-хромосомная сигнатура тестируемого в классическом ФТДНА-шном виде будет выглядеть следущим образом:
L269-:M201-:M379-:Page83+:L265-:Page29(M173+,P241+):M420+:Page7+.
Из чего еrgo (следует), что тестант принадлежит к группе R1a1a1























Для отправки комментария необходимо войти на сайт.