Начало февраля порадовало важным событием: на сервере Bioraxiv размещен препринт монументальной статьи Haak et al. (Iosif Lazaridis , Nick Patterson , Nadin Rohland , Swapan Mallick , Bastien Llamas , Guido Brandt , Susanne Nordenfelt , Eadaoin Harney , Kristin Stewardson , Qiaomei Fu , Alissa Mittnik , Eszter Bánffy , Christos Economou , Michael Francken , Susanne Friederich , Rafael Garrido Pena , Fredrik Hallgren , Valery Khartanovich , Aleksandr Khokhlov , Michael Kunst , Pavel Kuznetsov , Harald Meller , Oleg Mochalov , Vayacheslav Moiseyev , Nicole Nicklisch , Sandra L. Pichler , Roberto Risch , Manuel A. Rojo Guerra , Christina Roth , Anna Szécsényi-Nagy , Joachim Wahl , Matthias Meyer , Johannes Krause , Dorcas Brown , David Anthony , Alan Cooper , Kurt Werner Alt , David Reich) «Massive migration from the steppe is a source for Indo-European languages in Europe». Несмотря на то, что сама статья еще находится на стадии препринта, уже сейчас очевидна глубина проработки материала. Нет никаких сомнений в том, что это объемное, вдумчивое и тщательное исследование войдет в число главных работ в области индоевропеистики. По своей сути, коллектив авторов подвел в этой работе итоги всех предыдущих исследований введенных «полных геномов» древних жителей Европы (возрастом в 8 000 -4 000 лет), введенных в научных оборот за последние 2-3 года. Благодаря систематическому подходу к материалу и синтезу предыдущих наработок, а также за счет использования новейших физико-химических методов экстрагирования палео-ДНК вкупе с передовым современейшим биоинформатическим программным обеспечением, авторы смогли строго и скурпулезно подойти к одному из важнейших вопросов истории, лингвистики и археологии — к вопросу о происхождении индоевропейцев. К чести авторов, они признают, что даже после столь внушительного по своим размерам и качеству исследования, вопрос о первичном месте происхождения индоевропейцев остается открытым, и поэтому собственно обсуждение релевантности исследования палео-ДНК в свете существующих 4 главных теорий происхождения индоевропейцев занимает в работе относительно мало места (стр.134-139). Впрочем, вряд ли кто всерьез ожидал от этого исследования окончательного ответа на все вопросы индоевропеистики. Тем не менее, подробный анализ аутосомного генома, а также однородительских маркеров (митохондриального генома и Y-хромосомы) представителей целого ряда культур неолита, медногл и бронзового века восточной и западной Европы, и в особенности представителей Ямной культуры, дает новую подпитку вечному спору между сторонниками разных версий происхождения индоевропейцев (т.к. многие вслед за Гимбутас связывают ямную культуру с общностью протоиндоевропейцев).
Зато остальная часть этой 172-страничной работы настолько богата (насыщена) фактическим материалом, что любой, даже самый искушенный, читатель попгенетической литературы получит большое удовольствие от приобщения к плодам многолетней работы умнейших ученых. А работа, действительно, проделана огромная. Ученые воссоздали геномные данные 69 европейцев, живших между 8 000-3000 лет тому назад, за счет обогащения амплифицированных библиотек палео-ДНК. Эти библиотеки палео-ДНК они использовали для целевого отбора 394577 таргентных полиморфизмов (снипов) в панелях Affymetrix Human Origins. Обогащение именно этих специально отобранных таргентных позиций позволило снизить необходимые для анализа древней ДНК объемы секвенирования в среднем примерно в 250 (!) раз, что позволило авторам изучить на порядок больше лиц в сравнении с предыдущими исследованиями, и получить более полные знания о прошлом.

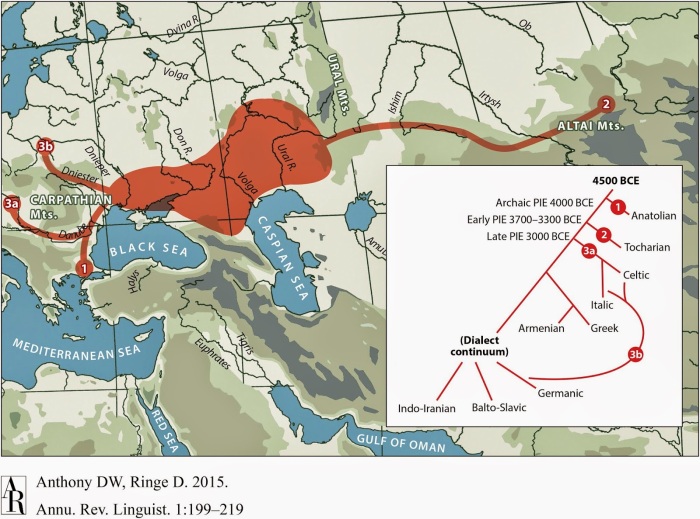
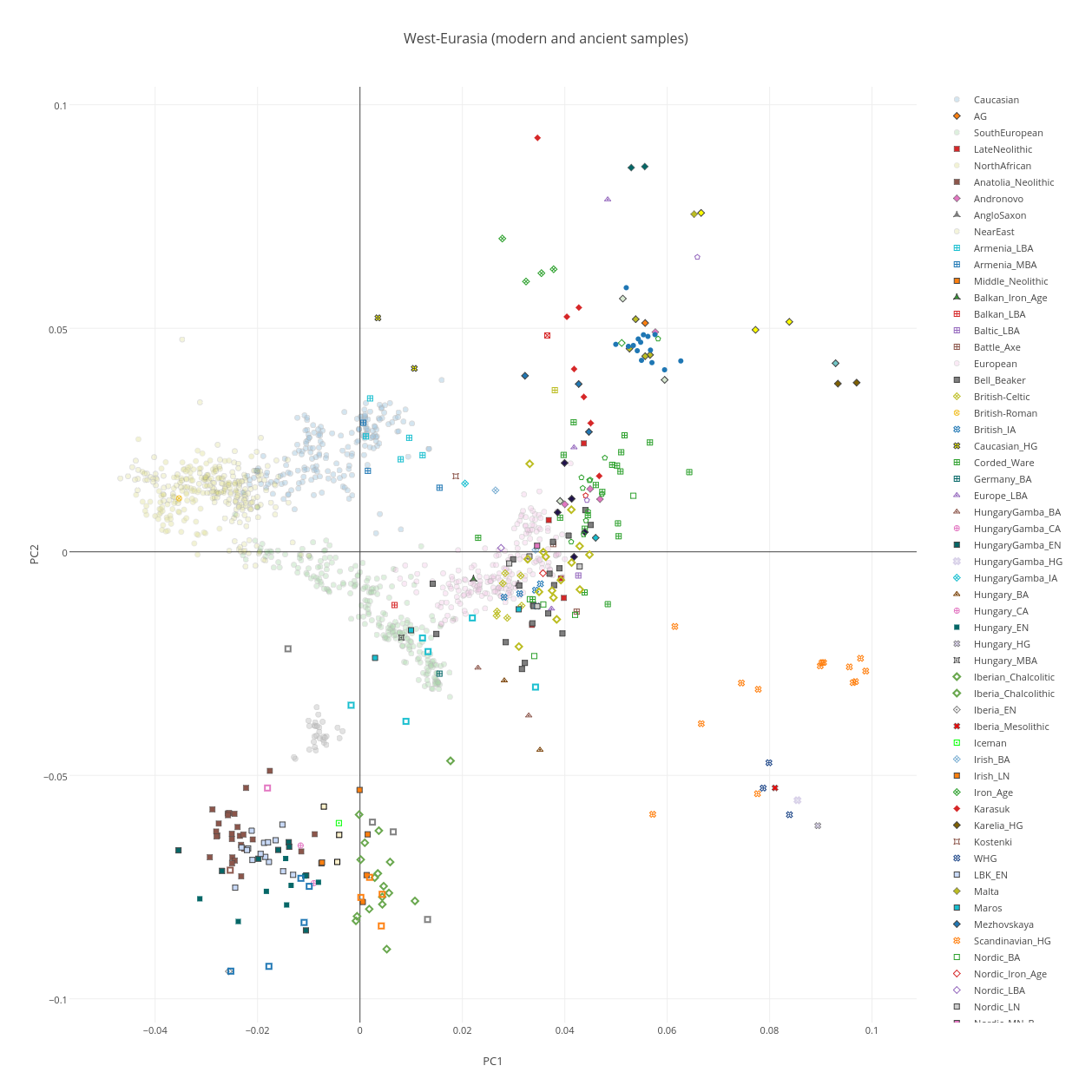
В работе показано, что уже 8,000-5,000 лет назад население западной и восточной Европы следовали противоположным траекториям развития.
На заре евпропейского неолита, примерно 8,000-7,000 лет назад, отдельныетесно связанные родством и отличные от коренных европейских охотников-собирателей, группы ранних земледельцев появились в Германии, Венгрии и Испании, в то время как Россия была населена особой группой восточных охотников-собирателей, имеющих родство с населением сибирского палеолита (24 000 л.н.в) , эта группа древних северо-евразийцев (ANE), представлена образцом MA1 (мальчик с палеолитической стоянки Malta-1 из южной Сибири); кроме того, этот компонент неплохо аппроксимируется «сибирской частью» генома изолированных индейцев Южной Америки (каритиана). Два образца охотников-собирателей из России (Карелия и Самары) образуют кластер «восточно-европейских охотников-собирателей «(EHG); пост-мезолитические охотники-собиратели из Люксембурга, Испании и Венгрии (WHG) находятся на противоположенном конце клина охотников-собирателей, в то время как охотники-собиратели Швеции (SHG) находятся посередине. Интересно отметить, что геном охотника-собирателя из Карелии представляет собой смесь 38-40% компонента ANE и 60-62% компонента WHG, причем величина ANE значительно выше 20% ANE, выведенных для шведского-охотника собирателя Motala-12 в предыдущем исследовании Lazaridis et al 2014).
В соответствии с тем, что EHG являются обмен население больше аллелей с «древних Северной евразийцев» (ГУ 7), чем любой другой.
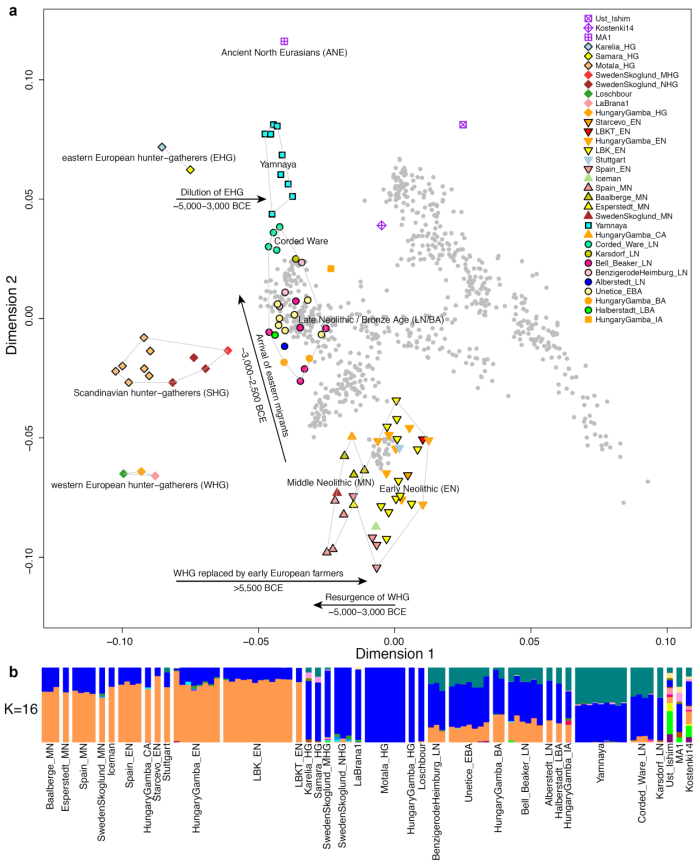
Примерно 6-5 тысяч лет назад, на большой части Европы назад произошел новый «подъем» потомков мезолитических охотников-собирателей, но в России степные скотоводы Ямной культуры время произошли от смешивания предыдущих племен восточно-европейских охотников-собирателей с населением ближневосточного происхождения. Население Ямной культуры отличалось от своих предшественников -восточно-европейских охотников-собирателей EHG — меньшим количеством обших аллелей с MA1 (| Z | = 6,7), что свидетельствует о процессе дисперсии носителей ANE на территории европейских степей где-то между 5 000-3 000 гг. до н.э. Уменьшение числа общих с образцом MA1 аллелей, вероятно объясняется появлением «примеси» от популяции или популяций, тесно связанных с
популяциями современного ближнего Востока, т.к. самая отрицательная f3-статистика (отрицательная статистика однозначно свидетельствует о примеси) наблюдается при моделировании жителей Ямной культуры как продукта смешивания носителей компонента EHG и современных популяций ближнего Востока, таких как армян (Z = -6,3).
Непосредственный контакт между населением западной и восточной Европой состоялся ~ 4500 лет назад, а в геноме поздне-неолитического населения культуры шнуровой керамики на территории Германии прослеживается 75%-ный «генетический» вклад «степного» компонента представителей ямной культуры. Таким образом геном жителей культуры шнуровой керамики «документирует» массовую миграцию населения с восточной периферии Европы в ее центральные области. Данный «степной» компонент Yamna (гибрид EHG и ближневосточных популяций) сохранялась в значительных пропорциях у всех имеюшисхя образцах из центральной Европы как минимум до ~ 3000 г.н.э, и повсеместно встречается у современных европейцев.
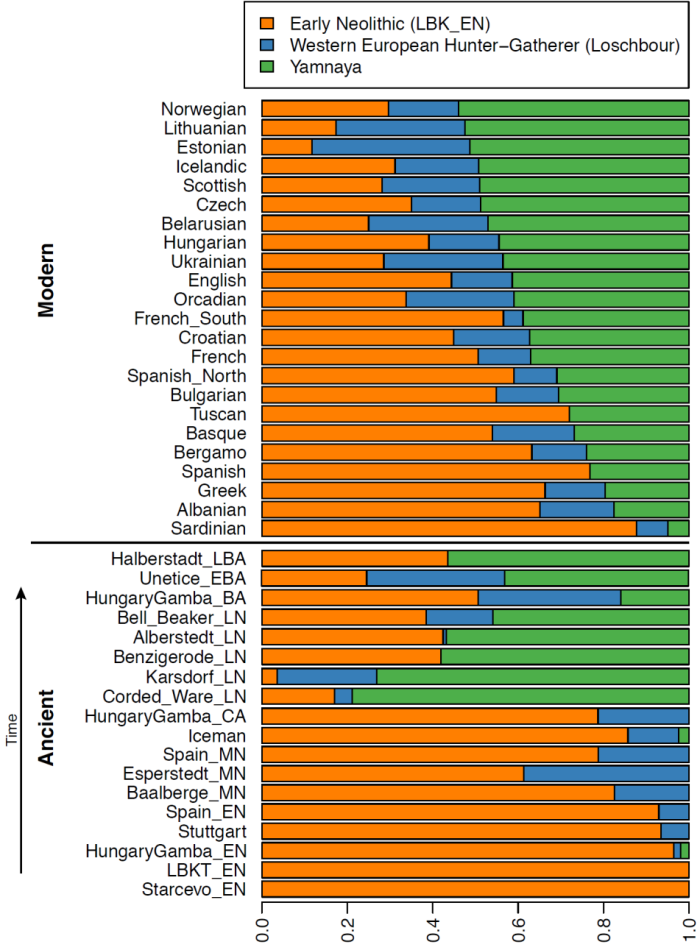
Если географическая дихотомия неолитического и мезолитического генетических компонентов в Европе была отмечена еще в работах пионеров популяционной генетки вроде Л. Кавалли-Сфорца, то данные этой работы позволяют вставить последнее звено в решении головоломки. На основании данных можно предположить, каким именно образом третий генетический компонент европейцев (ANE) попал из Сибири в Европу: сначала этот компонент несли представители EHG, затем он перешло к «ямникам» (смешанная популяция восточно-европейских охотников-собирателей и населения ближневосточного типа), а затем от ямников к представителям культуры шнуровой керамики, которые представляли собой смесь представителей ямной культуры с средне-неолитическими европейцами. В настоящее время этот компонент имеет плавное распределение на территории Европы, и по этой причине, мы можем использовать его в анализах структуры как генофонда целых народов, так и генома отдельно взятых людей.
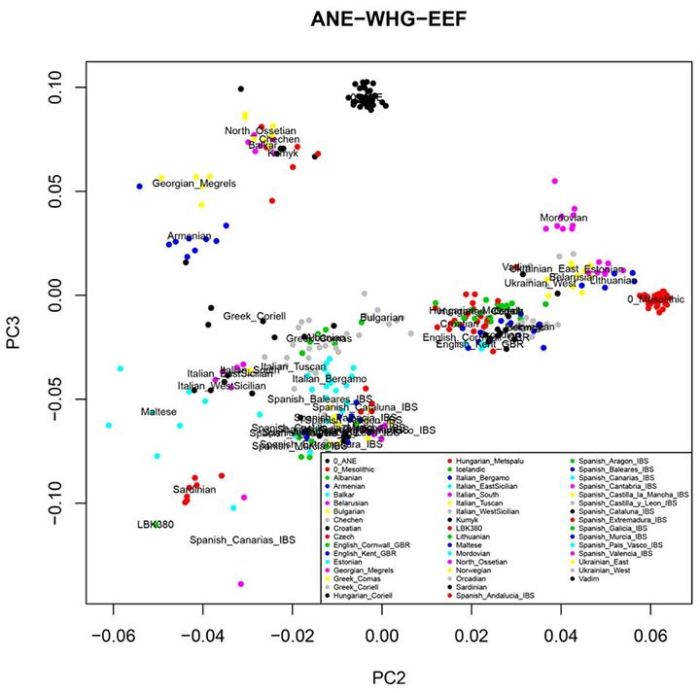
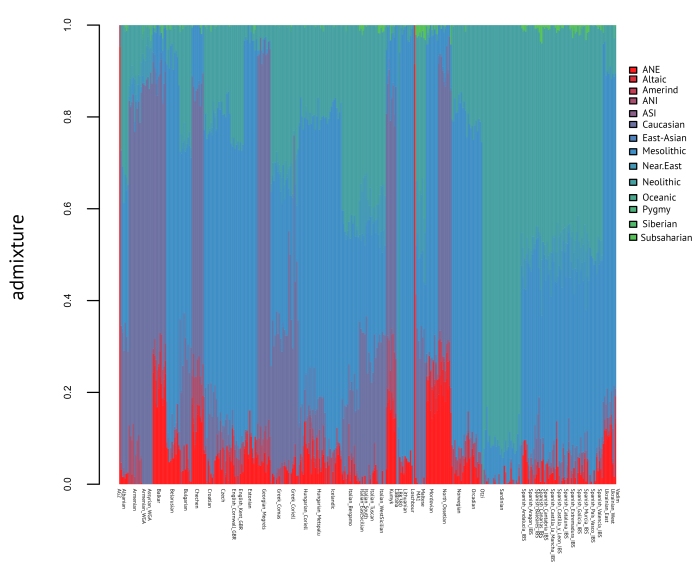
В январе я провел небольшой эксперимент c «чистым вариантом» аутосомного компонента ANE (ancestral North-Euarasians), впервые описаном в известной работе Lazaridis et al. 2014. Процедура вывода третьего генетического компонента в генофонде европейцев (ANE) оказалась гораздо сложнее, чем я предполагал в начале. Основная сложность заключалась в том, что в отличии от мезолитических и неолитческих сэмплов, у нас нет хорошого образца палеоДНК носителей компонента ANE. Пришлось заниматься реконструкцией генома, используя в качестве заменителей геном MA1 и не-восточноазиатскую часть генома индейцев каритиана. Технически, данный «компонент» был «синтезирован» в программе Plink с помощью 2последовательных запусков генерации «синтетических» популяций на основании частот аллелей аутосомных снипов, вычисленных в3 последовательных запусках программы Admixture.
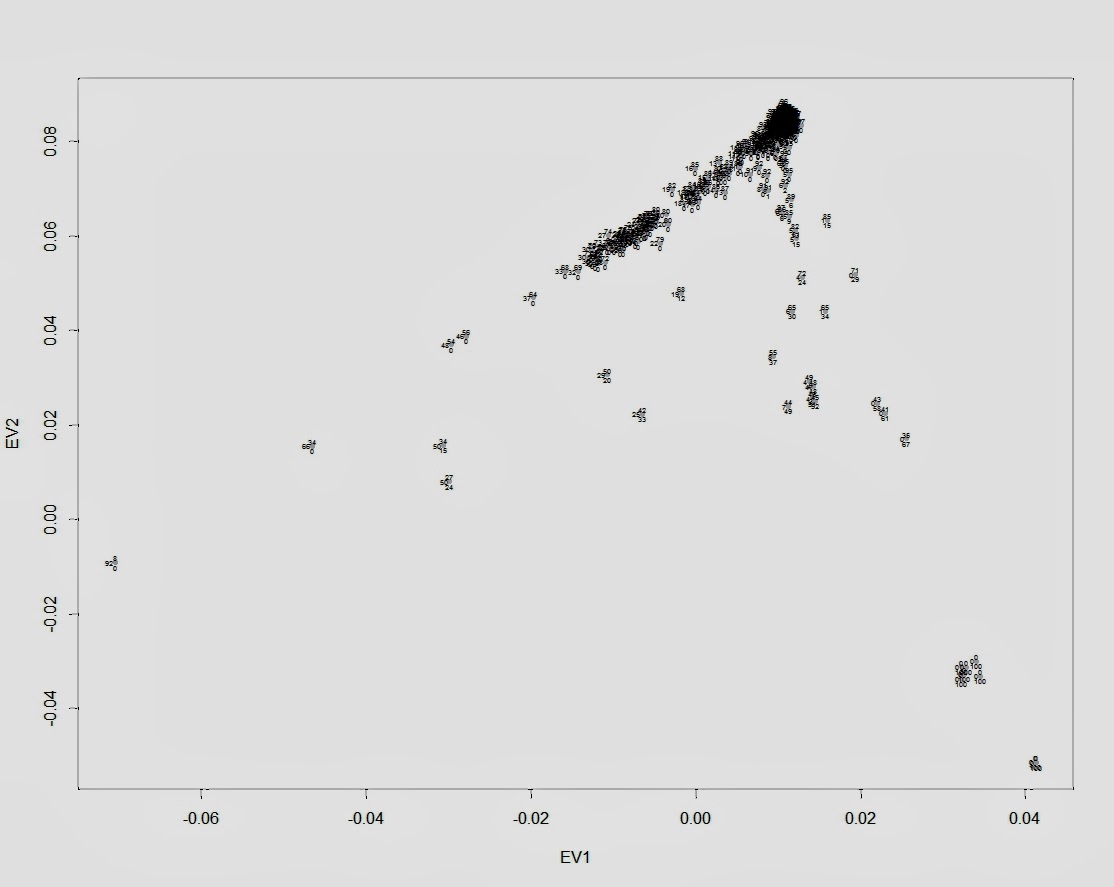
Я получил компонент с максимально приближенным значениями к значениям компонента ANE в разных популяциях мира в работе Lazaridis et al. 2014. Изучение этого компонента важно для понимания многих неясных моментов истоии древних популяций в восточной Европе и на северном Кавказе. Последние статьи и анонсы лаборатории Давида Рейха к новой статье о составляющих аутосомного генофонда представителей ямной культуры и культуры шнуровой керамики дают основания полагать, что компонент ANE в Евразии могли разносить потоки миграции индоевропейцев, а генетическое разнообразие жителей Европы и Кавказа практически вписывается внутри класссического треугольника (клинов) генетических компонентов ANE-WHG-EEF (см. ниже график PCA).
Формально,эта гипотеза проверяется с помощью инструментов f3-статистики (A; B,C) — формального теста на статистическую значимость предполагаемого варианта присутствия адмикса двух популяций-доноров в популяции-рецепиенте.
Я решил проверить надежность модели путем представления ряда европейских и кавказских популяций в виде продукта гибридизации носителей компонента ANE с «неолитическим» и «пост-мезолитическим» населением Европы (результаты ниже, Z в последней колонке.
| North-Caucas |
Caucasian |
ANE |
-0.0006748 |
5.13E-05 |
-13.166 |
| Mesolithic-North |
Loschbour |
ANE |
-0.0011573 |
0.0001605 |
-7.21171 |
| Mediterranean-Neolithic |
Otzi |
ANE |
-0.0012012 |
0.0002376 |
-5.05634 |
| Mesolithic-North |
LaBrana |
ANE |
-0.0010358 |
0.0002097 |
-4.94043 |
| Italian-East |
Otzi |
ANE |
-0.0012473 |
0.0005013 |
-2.48825 |
| Italian-East |
Italian-West |
ANE |
-0.0005022 |
0.0004325 |
-1.16129 |
| Maltese |
Otzi |
ANE |
-0.0001611 |
0.0004147 |
-0.388512 |
| Assyrian-1 |
Caucasian |
ANE |
-0.0002994 |
0.0009656 |
-0.310081 |
| Spanish-Canar |
Loschbour |
ANE |
-0.0002557 |
0.0011264 |
-0.227017 |
| Italian-East |
Maltese |
ANE |
2.36E-05 |
0.0003026 |
0.0779574 |
| Italian-East |
Caucasian |
ANE |
6.32E-05 |
0.000274 |
0.230808 |
| Spanish-Canar |
Otzi |
ANE |
0.0003307 |
0.0012476 |
0.265081 |
| Assyrian-1 |
Italian-West |
ANE |
0.0003321 |
0.0012207 |
0.272035 |
Практически все полученные варианты для современных популяций в тесте f3 дали отрицательную Z-оценку.
Буквой Z обозначается стандартная оценка, основанная на нормальном распределении. Иначе говоря, Z-o. является мерой отклонения от среднего, выраженной в единицах стандартного отклонения. Z –оценка будет иметь отрицательную величину, если показатели адмикса будут выше медианного значения.
Очень высокие или очень низкие (отрицательные) Z-оценки, связанные с очень маленькими p-значениями, располагаются в хвостах нормального распределения, и является значимыми, а не случайными. А значит, компонент ANE был индуцирован мною правильно.
Что еще любопытно, так это то, что третья составляющая современных европейцев — компонент ANE в моем эксперименте плавно разделился на две подсоставляющих — MA1 (древние сибиряки эпохи палеолита) и Кавказ (в качестве основы я брал геномы грузин и армян).
То есть, скорее всего компонент ANE появился в результате смешивания «труъ» древних северных евразийцев и кавказцев. Собственно, об этом намекал Рейх в анонсе своей публикации результатов анализа геномов жителей ямной культуры.
Вероятно, что кавказские популяции (особенно в Дагестане), характеризующиеся высоким уровнем гомо- и аутозиготности в определенном смысле «законсервировали» в своем геноме тот первый массовый вброс компонента ANE в свой генофонд. И по этой причине, например, без специальной методики, в программе Admixture практически весь компонент ANE маскируется бимодальным компонентом, вроде знаменитого Caucas-Gedrosia в одном из первых этно-популяционных калькуляторов проекта Dodecad. Похоже, что генетика может в очередной раз частично примирить две антиномные теории (вернее, целые кластеры теорий) происхождения ИЕ — анатолийскую и степную. Нечто подобное наблюдалось после прочтения геномов неандертальцев и получения убедительных фактов гибридизации предков соврменного человека и архаических гомининов — неандертальцев, денисовцев. В результате чего теории мультирегионального и монорегионального происхождения человка были хотя бы в отдельных моментах приведены к общему знаменателю.
Как я уже упоминал ранее, мой опыт с «выведением» предкового аутосомного компонента индоевропейцев полностью удался. Поскольку всем очевидно, что этот компонент родственен «североиндийскому предковому компоненту» (ANI — обозначение из статьи Reich et al. 2009 и Moorjani et al 2011) о структуре генофонда индийских этнических групп), я взял 10 индийских этнических групп, имеющихся в кураторском наборе лаборатории Райха и проанализировал эту выборку в Admixture на пропорции вхождения их геномов в 2 априорно заданные кластеры. Первый кластер ANE был априорно задан 40 синтетическим индивидами, сгенерированными в программе Plink на основании расчитанных ранее частот аллелей «чистого» компонента ANE. В качестве дополнительного контрольного образца я использовал геном Malta1, т.к. он содержит в себе наивысшее содержание компонента ANE. Второй кластер был задан 4 индивидами Onge (одна из аборигенных народностей Андаманских островов). Как неоднократно указывалось в литературе, именно жители Андаманских островов являются самыми «чистыми» носителями т.н «южно-индийского» предкового компонента ASI (на континенте чистых носителей этого «компонента» не осталось, в том числе и среди популяций дравидов, ведда и мунда). После нескольких экспериментов по эвристическому методу проб и ошибок, я получил более или менее приемлимое разделение индивидов на 2 кластера, а затем вычислил частоты аллелей в каждом из этих кластеров. Любопытно, что в ходе опыта, удалось не только выделить компонент ANI, но и добиться неплохого уровня дискримнации между компонентом ANI, ANE, и благодаря этому, оба компонента могут быть включены в мой следующий этно-популяционный калькулятор.
Надежность компонентов я проверил на собственных данных. В рабочей модели калькулятора K14 удельное распределение этно-генографических компонентов моего генома выглядит следующим образом:
68.75% — европейский мезолитический компонент
13.12% — северо-евразийский компонент ANE
10.23% — европейский неолитический компонент
4% — ANI (северо-индийский предковый компонент)
1.6% — кавказский компонент
1.2% — алтайский компонент
0.2% — сибирский компонент




































































{kind=link}
Для отправки комментария необходимо войти на сайт.