На портале Генофонд.ру размещен реферат важной статьи, подводящей промежуточные итоги изучения древней ДНК. Я позволю себе удовольствие процитировать себе некоторые места этого замечательного обзора, написанного ув. Надеждой Марковой
Термин «древняя ДНК» возник в научной литературе в 1980-х годах в связи с появлением новой области исследований, которая получила название «молекулярная палеонтология». С развитием сначала методов ДНК-амплификации (полимеразной цепной реакции), а потом методов секвенирования нового поколения эта область получила мощный толчок к развитию и сегодня стала основным средством реконструкции эволюции живых организмов, и в том числе реконструкции истории человека.
Революция в эволюционной генетике
Исследование древней ДНК совершило революцию в эволюционной генетике, так как появилась возможность напрямую исследовать прошлое, законсервированное в «капсуле времени» ДНК, пишут авторы статьи. Работы последних десятилетий показали, что древняя ДНК может сохраняться в костях, зубах, мумифицированных и замороженных тканях, и может быть извлечена из этих древних образцов. Впервые древняя ДНК была извлечена в 1984 г. (Higuchi et al.) из высохшей мышцы вымершего родственника зебры. Но ее анализ целиком зависел от развития технологий, поэтому стал возможен с появлением ДНК-амплификации (метод полимеразно-цепной реакции – ПЦР), и вышел на новый уровень с появлением методов секвенирования нового поколения. На рисунке авторы представили основные вехи в истории изучения древней ДНК.
О методологии исследования палео-ДНК
Методы палеогенетики оказались незаменимы, чтобы разобраться в ключевых этапах человеческой цивилизации. Например, понять, как именно происходила смена обществ охотников-собирателей на первых земледельцев, как распространялось по Европе сельское хозяйство – имела ли место передача технологий от одних популяций другим или же происходила смена самих популяций («циркуляция идей или людей»). Анализ древней ДНК показал, что между периодами 8 и 5 тысяч лет назад Европа не была генетически однородной: первые земледельцы с Ближнего Востока мигрировали в Западную Европу и смешивались там с местными охотниками-собирателями. В Восточную Европу около 6-5 тыс. лет назад туда пришли группы людей из Анатолии, которые смешавшись с охотниками-собирателями, дали начало популяциям скотоводов, наиболее успешная из которых известна по ямной культуре. Полагают, что именно миграции ямников из понто-каспийских степей на запад и на восток около 4,5 тыс. лет назад можно связать с распространением технологий и, возможно, языков индоевропейской семьи.
Древняя ДНК может помочь и в изучении развития признаков, характерных только для Homosapiens, таких как речь, подчеркивают авторы статьи. Изучение генетических вариаций, связанных с языком, дает информацию о том, когда мог возникнуть сложный язык, присущий человеку. Так, было показано, что определенный вариант гена FOXP2 (именно его в первую очередь связывают с развитием речи) имелся уже у неандертальцев. Вероятно, считают специалисты, этот вариант возник у общих предков неандертальцев и современного человека.
Древняя ДНК помогает в изучении адаптации человека к разным условиям среды. При анализе древних геномов в них были выявлены сигналы отбора, связанных с изменением диеты, чувствительностью к ультрафиолету и пр. Так, становится ясно, как распространялись по Европе такие черты, как светлая кожа и толерантность к лактозе (способность переваривать молоко во взрослом возрасте).
Трудности в изучении палео-ДНК и их преодоление
Одна из основных проблем, с которыми сталкиваются исследователи древней ДНК, это ее деградация, которая неизбежно происходит со временем. Обычно ДНК из древних образцов сильно фрагментирована, загрязнена микробной ДНК и химически модифицирована. Причем степень деградации в больше степени зависит от условий, в которых находился древних образец (температура, влажность), чем от его возраста. Последние исследования показали, что теоретический предел возраста образца, из которого можно извлечь ДНК, составляет 1-1,5 млн лет. Авторы описывают методы, которыми можно преодолеть трудности, связанные с особенностями древней ДНК.
Фрагментация ДНК может быть частично преодолена с помощью современных протоколов, позволяющих извлекать и анализировать очень короткие фрагменты, длиной 50-70 нуклеотидов. К тому же, методы секвенирования нового поколения ориентированы на анализ коротких фрагментов, длина которых составляет 50-100 нуклеотидов.
Большую проблему составляет контаминация древней ДНК современной ДНК. Преодолеть ее нужно путем строгого соблюдения протоколов, учитывающих правила сбора образов, обработки рабочих помещений, применение методов ДНК-аутентификации, независимой перепроверки результатов и пр. Развиваются также методы механической и химической деконтаминации – авторы их описывают.
Еще одна важная проблема – посмертное изменение ДНК из-за гидролиза и окисления, вызывающее деаминацию нуклеотидов, которая ведет к ложным результатам ПЦР. Авторы описывают несколько молекулярно-генетических и биоинформатичесих подходов для преодоления этой проблемы, с ними можно ознакомиться в тексте статьи.
Инструменты анализа
С увеличением числа образцов древней ДНК ученые получают возможность исследовать древнюю генетическую изменчивость на популяционном уровне и сравнивать ее с современной. Различные методы (PCA, STRUCTURE, ADMIXTURE, SPAMIX, SPA, ADMIXTOOLS, GPS, LAMP, HAPMIX, reAdmix, MUTLIMIX, mSpectrum, SABER и др.), которые были разработаны для анализа современных популяций, применяются и к древним популяциям. В комбинации с антропологическими данными и историческими сведениями они позволяют реконструировать пути миграций, определять состав предков той или иной популяции, выяснять географическое происхождение гаплотипов.
Эпигенетика и палео-ДНК
Фенотипическое проявление генотипической изменчивости зависит не только от изменчивости тех или иных аллелей в геноме, но и от степени экспрессии генов, а она во многом определяется химическими модификациями, не затрагивающими последовательность нуклеотидов в ДНК, то есть эпигенетическими. Это метилирование ДНК, модификация белков-гистонов, спектр некодирующей РНК. Последние исследования показали, что некоторые эпигенетические модификации сохраняются и postmortem. Так, удалось картировать метилирование генома неандертальцев и денисовцев. Выяснилось, что некоторые гены были более метилированы у древних людей, чем у современных. Анализ метилирования позволяет также определить возраст индивида (как современного – что важно для криминалистики, так и древнего).
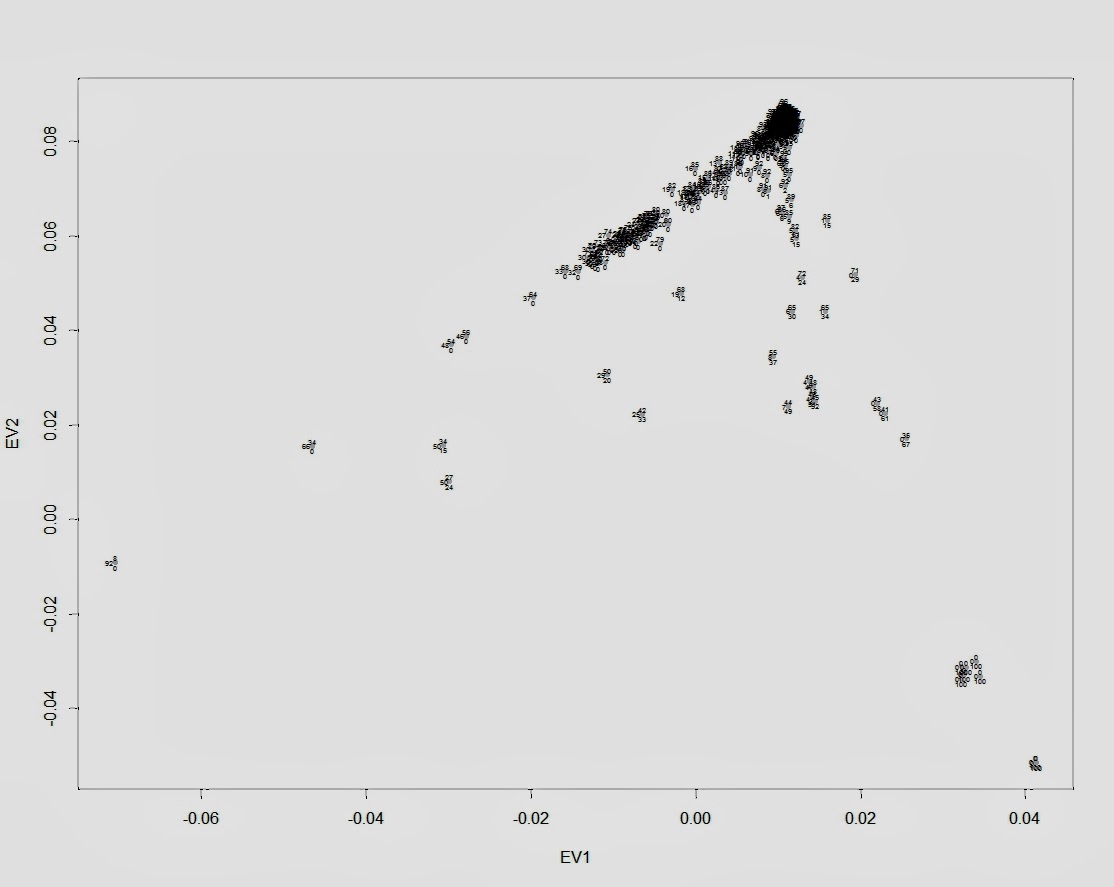
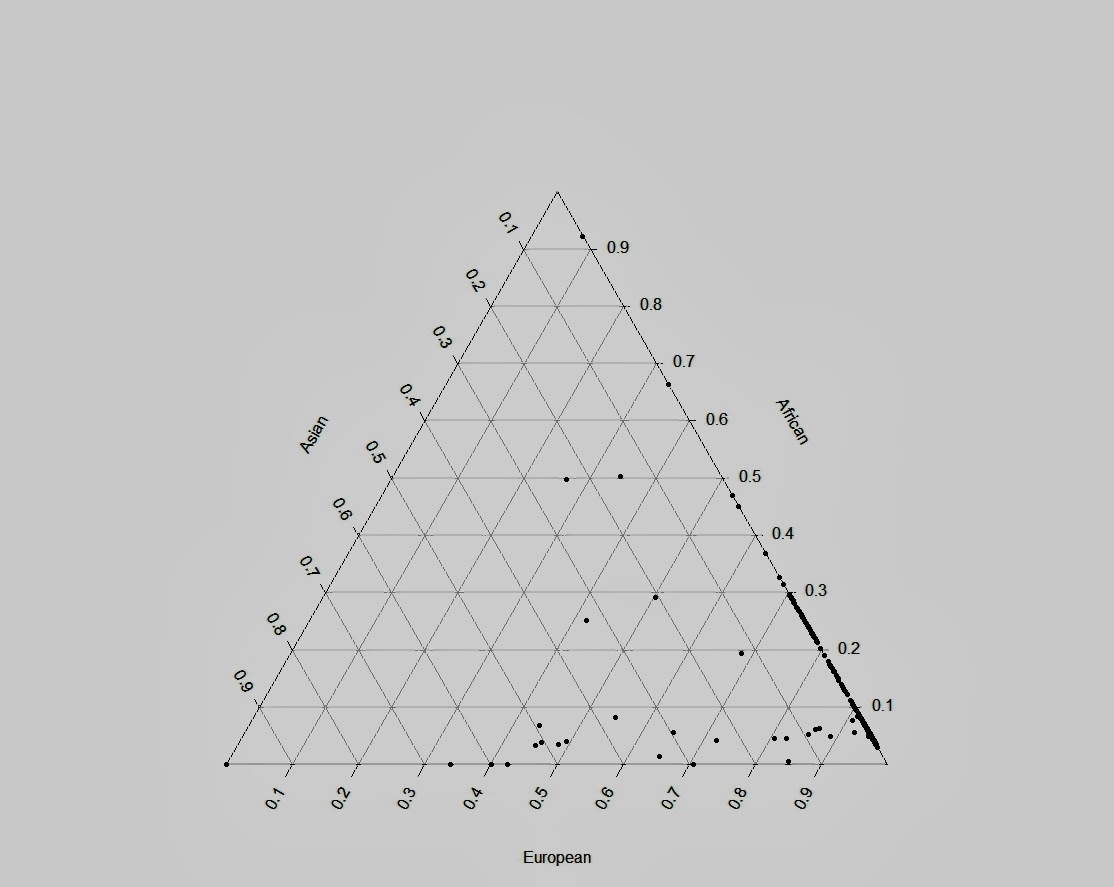
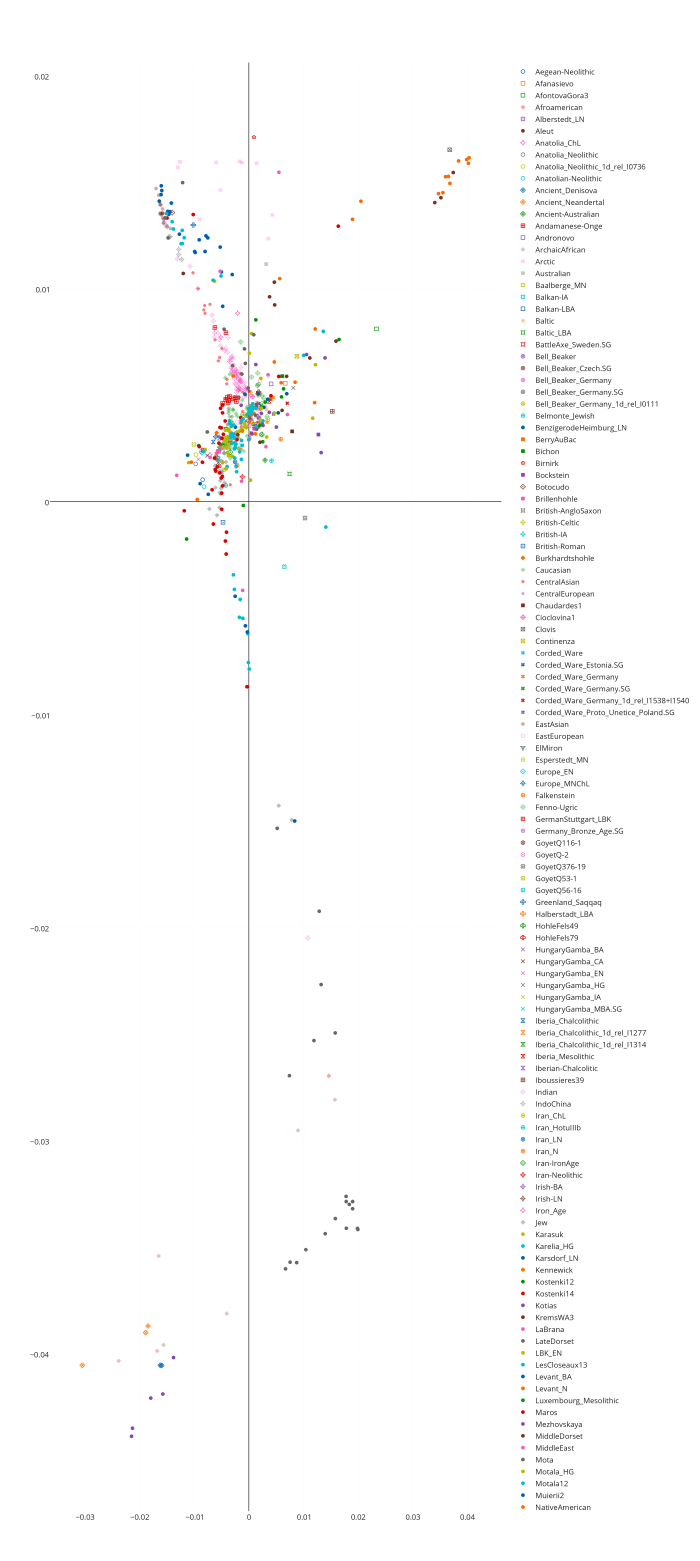

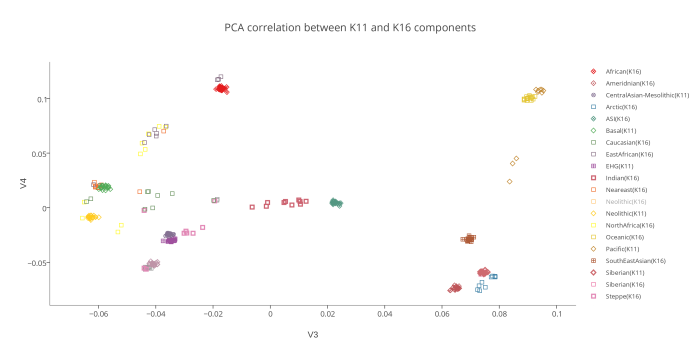 Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).
Вот эта таблица с усредненными значениями «симулянтов» компонентов K16 в калькуляторе K11 (колонки — компоненты K16, столбцы — компоненты K11, их пересечения — проекция компонентов K16 в компоненты K11).

















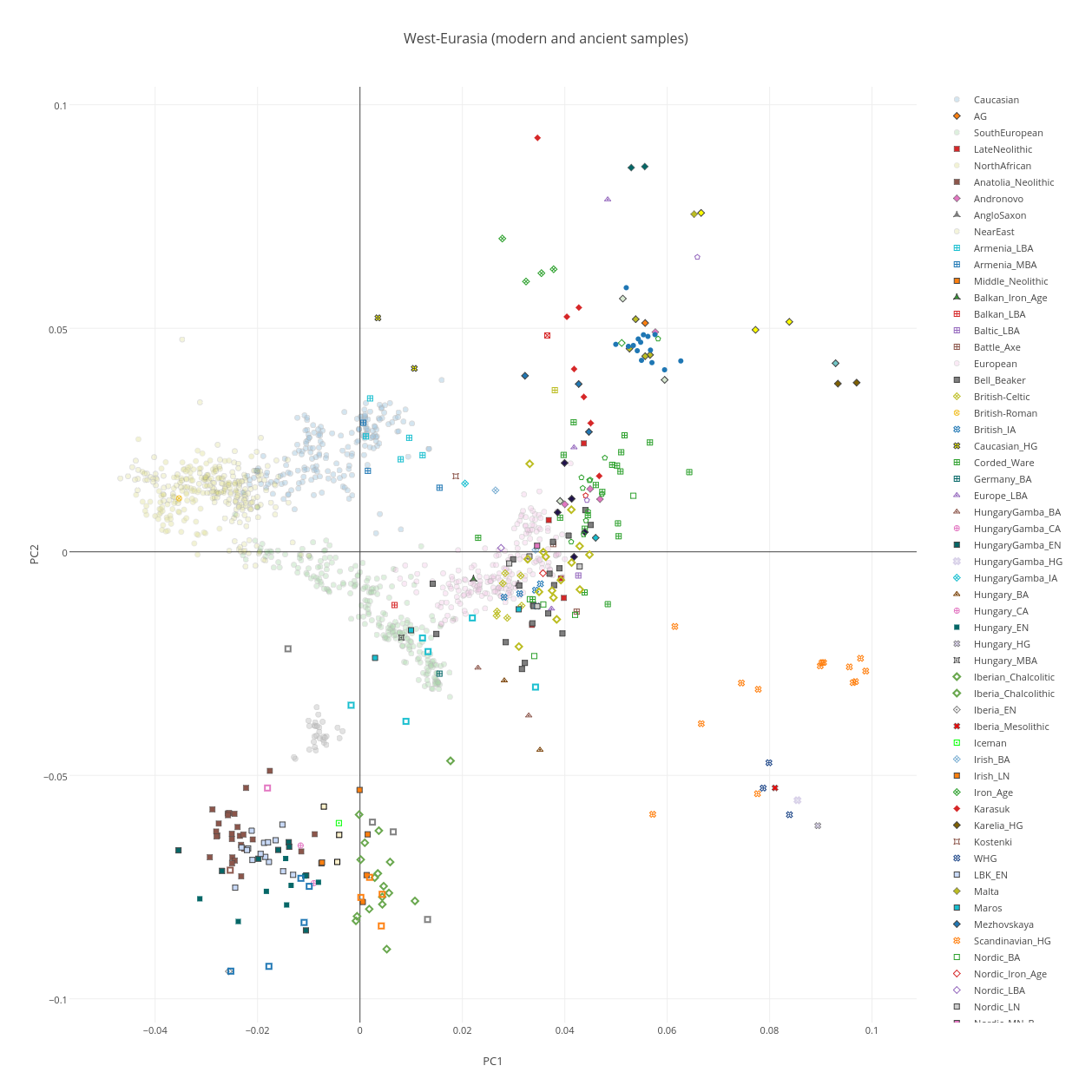








Для отправки комментария необходимо войти на сайт.